Genome-Wide Identification and Characterization of Point Mutations in the SARS-CoV-2 Genome
Article information

Abstract
Objectives
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged in Wuhan, China, in December 2019 and has been rapidly spreading worldwide. Although the causal relationship among mutations and the features of SARS-CoV-2 such as rapid transmission, pathogenicity, and tropism, remains unclear, our results of genomic mutations in SARS-CoV-2 may help to interpret the interaction between genomic characterization in SARS-CoV-2 and infectivity with the host.
Methods
A total of 4,254 genomic sequences of SARS-CoV-2 were collected from the Global Initiative on Sharing all Influenza Data (GISAID). Multiple sequence alignment for phylogenetic analysis and comparative genomic approach for mutation analysis were conducted using Molecular Evolutionary Genetics Analysis (MEGA), and an in-house program based on Perl language, respectively.
Results
Phylogenetic analysis of SARS-CoV-2 strains indicated that there were 3 major clades including S, V, and G, and 2 subclades (G.1 and G.2). There were 767 types of synonymous and 1,352 types of non-synonymous mutation. ORF1a, ORF1b, S, and N genes were detected at high frequency, whereas ORF7b and E genes exhibited low frequency. In the receptor-binding domain (RBD) of the S gene, 11 non-synonymous mutations were observed in the region adjacent to the angiotensin converting enzyme 2 (ACE2) binding site.
Conclusion
It has been reported that the rapid infectivity and transmission of SARS-CoV-2 associated with host receptor affinity are derived from several mutations in its genes. Without these genetic mutations to enhance evolutionary adaptation, species recognition, host receptor affinity, and pathogenicity, it would not survive. It is expected that our results could provide an important clue in understanding the genomic characteristics of SARS-CoV-2.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged in Wuhan, China, in late December 2019. Since then, it has rapidly spread across the world, and was finally declared as a Public Health Emergency of International Concern (PHEIC) by the World Health Organization on January 30th, 2020 [1].
SARS-CoV-2 is taxonomically classified under Nidovirales order, Coronaviridae family, Coronavirinae subfamily, and betacornoavirus genus. It is an enveloped virus with nonsegmented, positive-sense, single-stranded RNA. Although SARS-CoV-2 presents with a lower pathogenicity than severe acute respiratory syndrome coronavirus (SARS-CoV) which emerged in 2002–2003, and Middle-East respiratory syndrome coronavirus (MERS-CoV) which emerged in 2012, it reveals more rapidly human-to-human transmission [2].
The genome of SARS-CoV-2 consists of non-segmented RNA that includes a 5’ untranslated region (UTR), structural proteins, non-structural proteins, several accessory proteins (open reading frames), and a 3’ UTR. The ORF1ab of several ORFs is proteolytically cleaved into 16 putative non-structural proteins (nsp1-16) for genome maintenance and replicase complex formation in viral replication. The structural proteins essential in viral particles include the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins. The receptor-binding domain (RBD) of the S protein is crucial for binding directly to the human receptor ACE2, inducing viral entry, and determining host tropism and transmission capacity [3-5]. The S protein is cleaved into 2 subunits (S1 and S2). The S1 subunit directly recognizes and attaches to human receptor ACE2, while S2 fuses the host cell membrane with viral membranes allowing entry of SARS-CoV-2 [6]. In general, RNA viruses like SARS-CoV-2 undergo rapid mutation, enabling evolutionary and genetic diversity which result in alterations such as viral transmissibility, receptor affinity, host tropism, and pathogenicity.
In recent years, several studies based on mutation analysis of SARS-CoV-2 genome have attempted to understand phylogenetic relationships, host infectivity, human-to-human transmission, viral tropism, and pathogenicity of SARS-CoV in humans. Firstly, the comparative evolutionary diversity in point mutations (synonymous-non-synonymous mutations) are suggestive that SARS-CoV-2 should to be classified into 3 major clades (S, G, and V) and other clades according to amino acid changes [7-9]. Secondly, the high affinity and stable structure of RBD/ACE2 have been associated with amino acid variations in the RBD such as the high affinity group (N354D, D364Y, V367F, and W436R) [10], and the high ACE2-binding affinity and stability group (484-NGVEGFN-490, Q496N, and Q496Y) [11]. Thirdly, the deletion of 382 nucleotides towards the 3’ end of the viral genome may have an impact on viral phenotype [12], and the QTQTN motif adjacent to the polybasic cleavage site (RRAR, chain of amino acids) at the bridge between S1 and S2 may be related to host adaptation [13]. In addition, insertion of the RRAR which has been well known to determine high or low pathogenicity in avian influenza virus may be important in determining transmissibility and pathogenesis of SARS-CoV-2 [14]. Finally, primer-template mismatch has been known to affect the stability and functionality of polymerase. In particular, the primer-template mismatch located in the primer 3’ end region can interfere with polymerase active sites, and this may have a significant impact on the accuracy of the molecular diagnosis using primers or probes [15].
Therefore, we analyzed the mutations of the SARS-CoV-2 genome by focusing on phylogenetic evolution, RBD region, deletion mutations in polybasic cleavage site, and primer-template mismatches in the genome. Although the mechanisms responsible for rapid transmission, pathogenicity, and tropism in SARS-CoV-2 remain unclear, identification of mutations in the SARS-CoV-2 genome may help to interpret the high infectivity of the virus with the host.
Materials and Methods
The set of 4,254 SARS-CoV-2 genome sequences and acknowledgment files were downloaded from the EpiCoV browser (https://epicov.org/epi3) of the GISAID [16]. The raw data were processed by removing unnecessary genome sequences with low-quality reads, base calling errors, unsolved nucleotides as “N”, and small gaps. To investigate the genomewide phylogenetic analysis, we recombined 12 coding sequences (ORF1a, ORF1b, S, M, E, N, ORF3, ORF6, ORF7a, ORF7b, ORF8, and ORF10), excluding 5′ and 3′ UTR, low-quality sequences, and strains with high sequence similarity within the same clade. As a result of the processing, a reference genome (hCoV-19/Wuhan-Hu-1/2019, EPI_ISL_402125, 29,903 bp) and 178 fully complete genomes were collected. Phylogenetic analysis was performed to identify evolutionary relationships across the genome by using the MEGA [17] with parameters such as neighbor joining method, bootstrap 1,000 replications for the phylogeny test, Kimura 2-parameter as a substitution model, and pairwise deletion as gap/missing data treatment. For identifying the types of point mutation (synonymous and non-synonymous mutation) from 4,254 sequences, we separately extracted 12 fully coding sequences (CDSs) determining the sequence of amino acids in a protein from the genomes, and then conducted an in-house program based on Perl computer programming language to analyze mutations among the CDSs and the reference genome. In nomenclature for the replacement from one amino acid to a different amino acid in a gene, we illustrated it as the gene name surrounded by parenthesis after the substitution of amino acids. For example, D614G (S) means that an aspartic acid is converted into a glycine at amino acid position 614 of the S gene in comparison with the reference strain (hCoV-19/Wuhan-Hu-1/2019). For the comparative analysis of primer- and probe-template mismatches, we referred several lists published on the of Centers for Disease Control and Prevention (CDC) websites; https://www.who.int/docs/default-source/coronaviruse/uscdcrt-pcr-panel-primer-probes.pdf in CDC Atlanta, https://www.who.int/docs/default-source/coronaviruse/protocol-v2-1.pdf of the Charité Virology in Germany, https://www.who.int/docs/default-source/coronaviruse/peiris-protocol-16-1-20.pdf in school of public health at the university of Hong Kong, and https://www.who.int/docs/default-source/coronaviruse/real-time-rt-pcr-assays-for-the-detection-of-sars-cov-2-institut-pasteur-paris.pdf of the Institut Pasteur in Paris (Table 1).

Primers and probes for the detection of SARS-CoV-2 of global research institutions.
Results
1. Phylogenetic analysis of SARS-CoV-2 population
Phylogenetic analysis was carried out with 178 representative strains to comprehend monophyletic distribution of the SARS-CoV-2 population. The phylogenetic tree showed 3 major clades (S, G, and V clades), similar to previous reports by GISAID. These clades (S, G, and V clades) were determined by the mutations [L84S (ORF8), G251V (ORF3a), and D614G (S)], respectively (Figure 1). Of the three mutations determining the clades, the significant D614G(S) in G clade is located in the adjacent polybasic cleavage site, but its actual function on RDB/ACE2 affinity is unclear. In addition to the three clades, two subclades belonging G clade were observed and they were named G.1, and G.2 clades referring to their parent clade name. The G.1 and G.2 subclades were determined by three mutations [G204R (N), R203K (N), and P214L (ORF1b)], and one mutation [Q57H (ORF3a)], respectively (Figure 2). In this respect, the derivation of G.1 and G.2 subclades from the G clade shows that a clade can be also determined by one or more mutations.
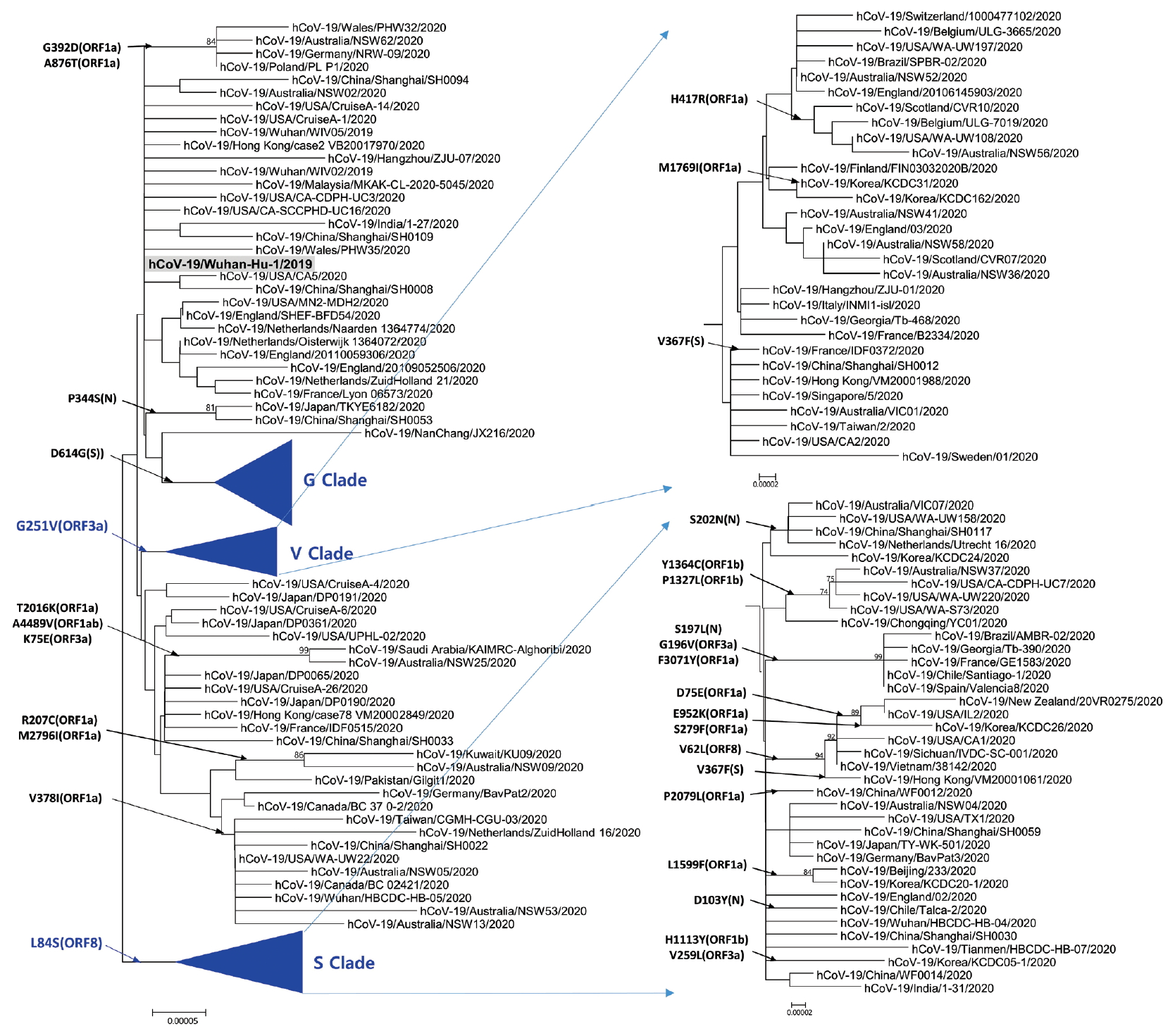
Phylogenetic tree of SARS-CoV-2. This tree was performed by using the MEGA with parameters such as neighbor joining method, bootstrap 1,000 replications for the phylogeny test, Kimura 2-parameter for substitution model, and pairwise deletion for gap/missing data treatment. S and V clades determined by L84G (ORF8) and G251V (ORF3a), respectively. The reference strain (hCoV-19/Wuhan-Hu-1/2019) is indicated by bold letters on a light gray background. The notation of amino acid substitutions used here means replacements from amino acid of the reference strain on left to a difference amino acid of the corresponding strain on right. MEGA = molecular evolutionary genetics analysis.
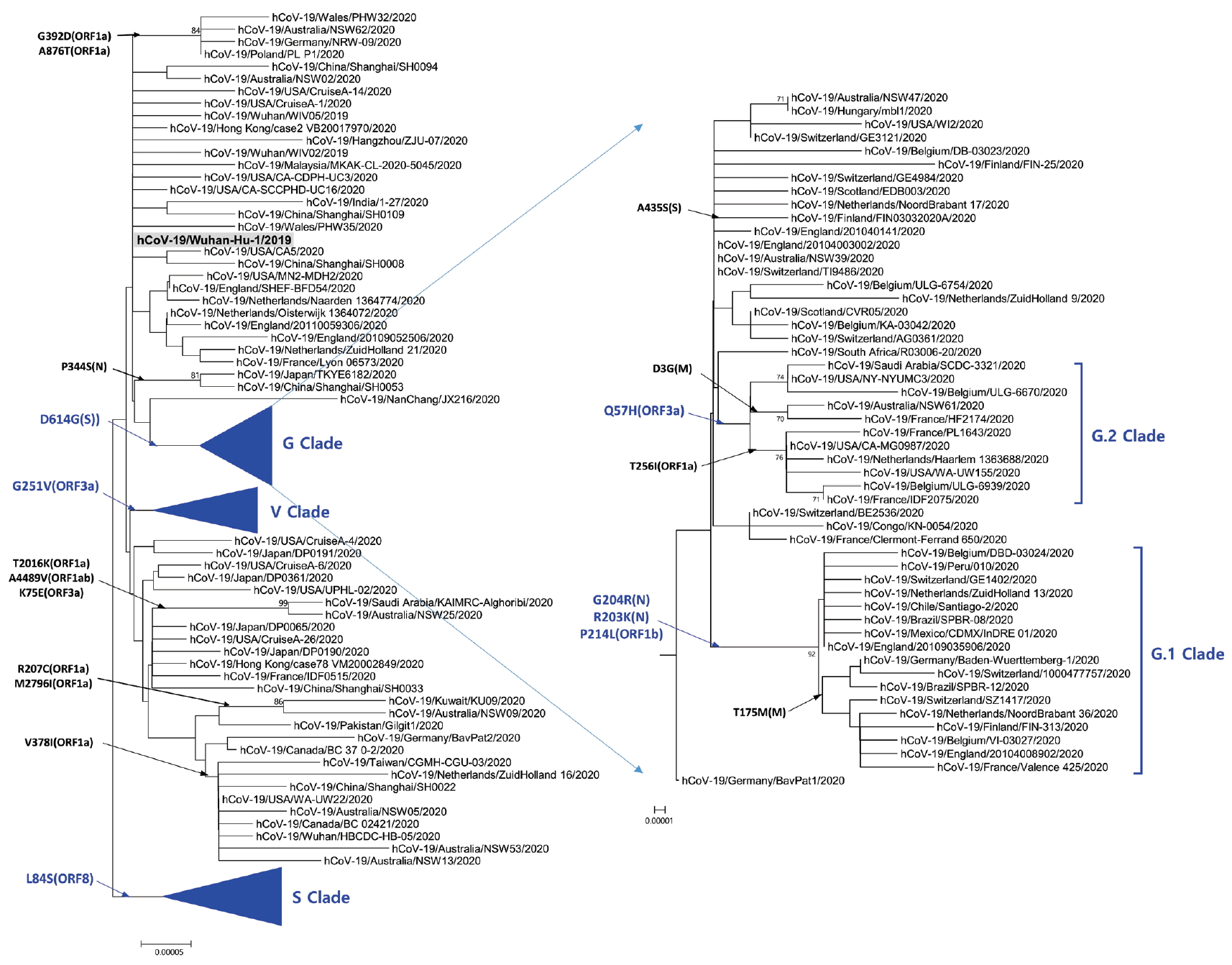
Phylogenetic tree of G clade and its subclades. G Clade determined by D614G (S) was classified into G.1 clade, G.2 clade, and other strains. G.1 and G.2 clades share Q57H (ORF3a) and G204R (N), R203K (N), together with P214L (ORF1b), respectively. The reference strain (hCoV-19/Wuhan-Hu-1/2019) is indicated by bold letters on a light gray background. The notation of amino acid substitutions used here means replacements from amino acid of the reference strain on left to a difference amino acid of the corresponding strain on right.
2. Frequency and types of mutation in SARS-CoV-2 genomes
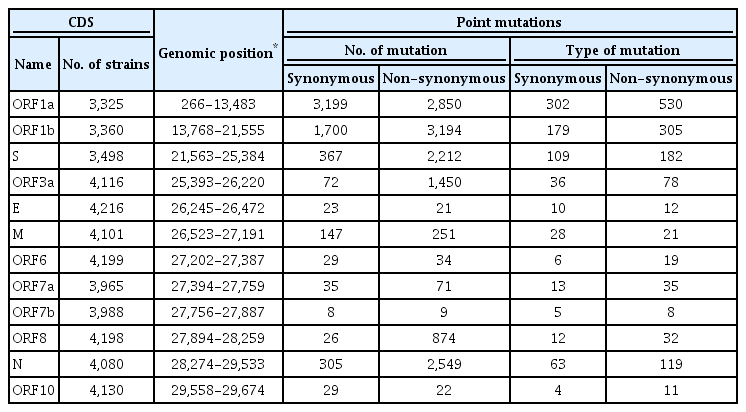
To identify the number and the types of mutation across a total of SARS-CoV-2 strains, the 12 different types of CDS from the 4,254 strains were completely extracted in accordance with the genomic positions presented by the NCBI’s GenBank format file (sequence ID: NC_045512.2). The unsuitable sequences with base calling errors, unsolved nucleotides as “N”, and undefinable gaps were excluded. A total of 47,176 CDSs were gathered from ORF1a, ORF1b, S, M, E, N, ORF3, ORF6, ORF7a, ORF7b, ORF8, and ORF10. We identified 767 types of synonymous and 1,352 types of non-synonymous mutation from them. Genes with high frequency mutations were ORF1a, ORF1b, S, N, and ORF3a. ORF1a showed the highest frequency mutations containing 302 types of synonymous and 530 types of non-synonymous mutation (Table 2, Figure 3). The non-synonymous mutations with high frequency in the genes were representative mutations in the clades: L84S (ORF8) in S clade, D614G (S) in G clade, G251V (ORF3a) in V clade, P214L (ORF1b), R203K (N) and G204R (N) in G.1 clade, and Q57H (ORF3a) in G.2 clade (Figures 4 and 5).
Frequency of point mutations by 12 coding sequences.
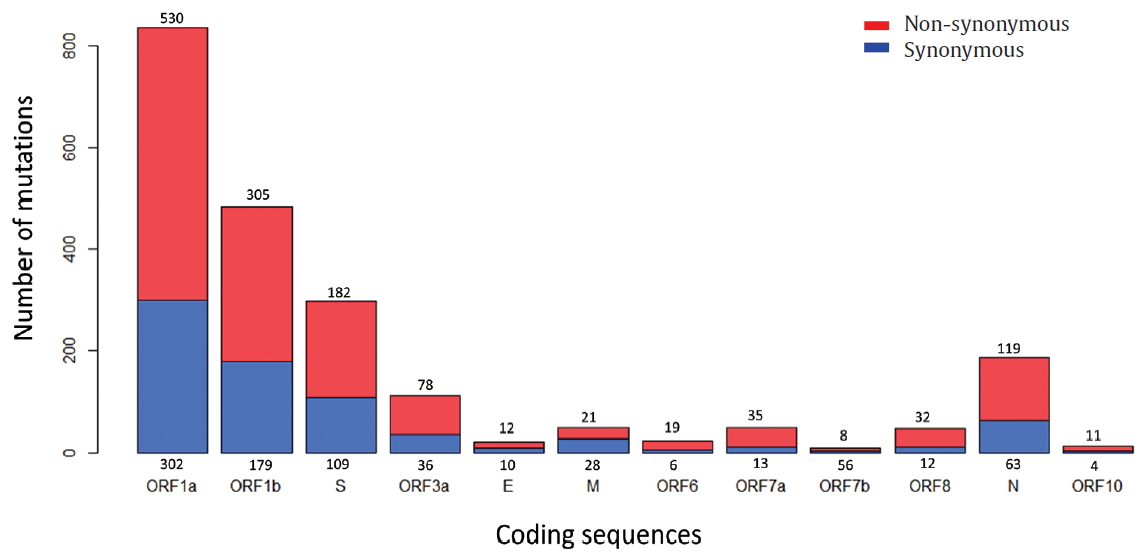
Types of mutation distribution in 12 coding sequences. This figure summarizes the distribution of point mutations on 12 coding sequences (ORF1a, ORF1b, S, ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N, and ORF10). Bars filled with red color indicates non-synonymous mutations and bars filled with blue color indicates synonymous mutation. Number above and below bars show the type of mutation.
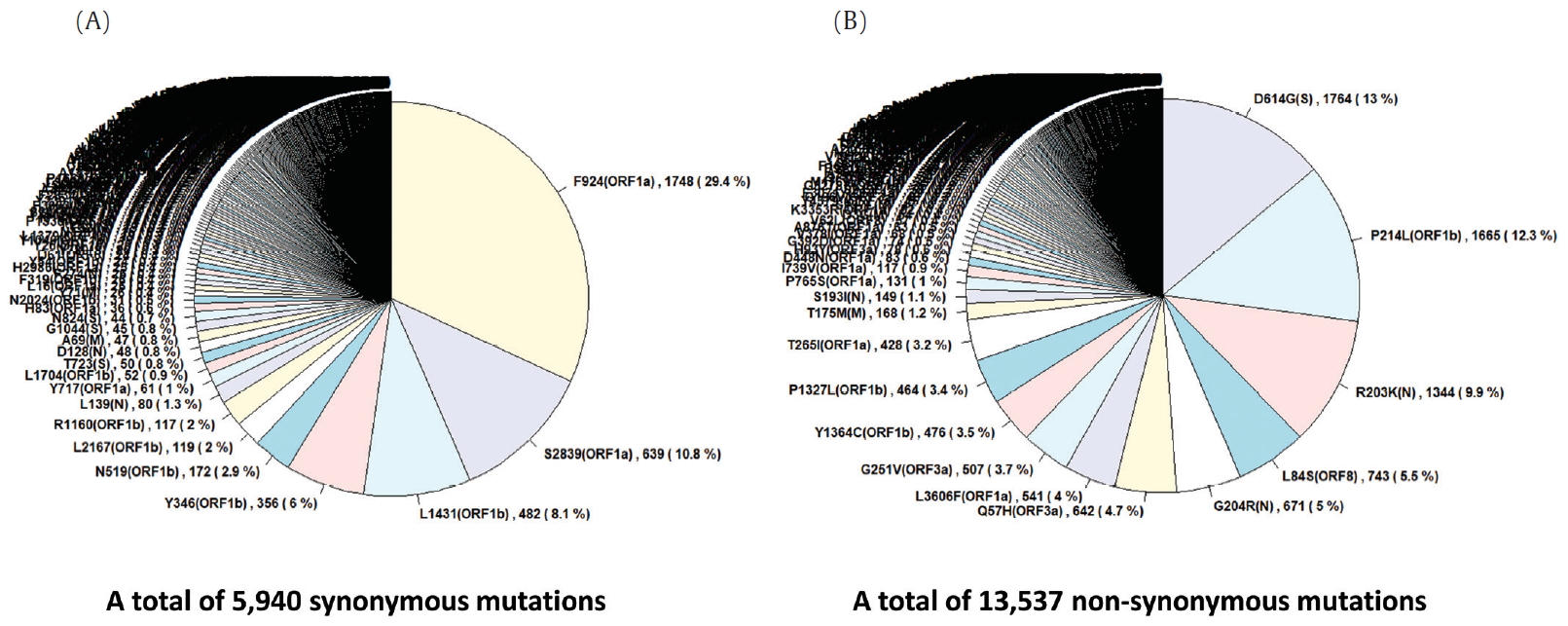
Frequency of mutations in coding sequences. (A) indicates frequency of 5,940 synonymous mutations (767 types of synonymous mutation) in 47,176 coding sequences from 4,254 strains. (B) indicates frequency of 13,537 non-synonymous mutations (1,352 types of non-synonymous mutation) in them.
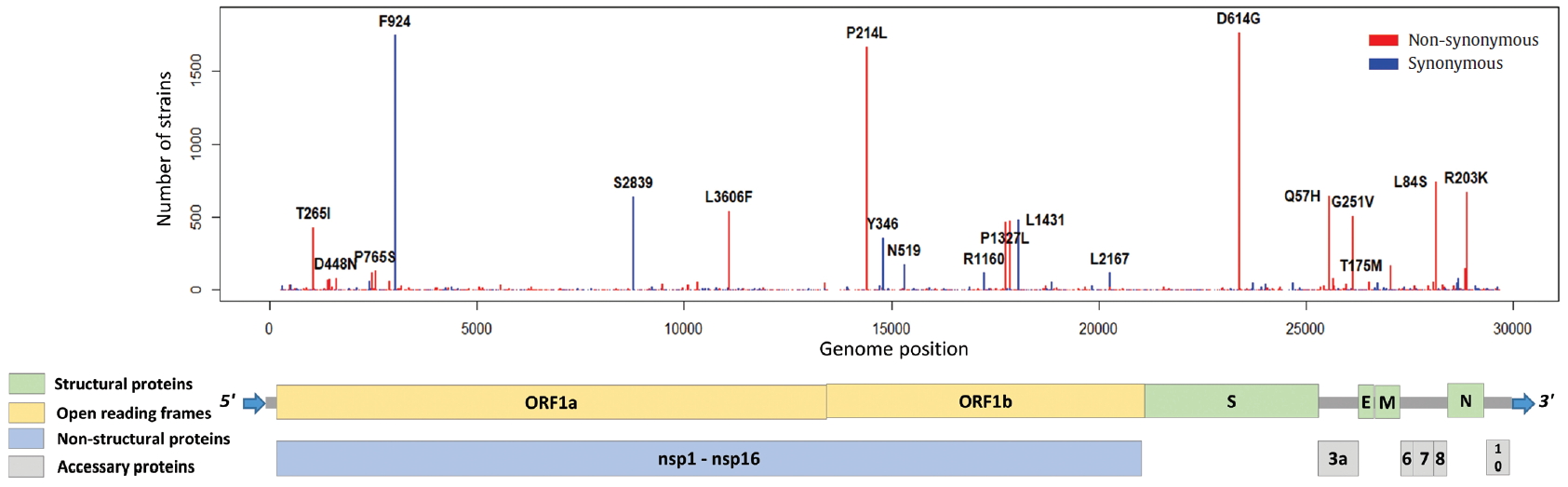
Distribution of mutations in SARS-CoV-2 whole genome. The length of SARS-CoV-2 genome is 29,903 bases. SARS-CoV-2 genome is composed of 5’ UTR, ORF1a, ORF1b, S, E, M, N, accessary proteins (ORF3a, ORF6, ORF7, ORF8, and ORF10), and 3’ UTR. The ORF1a and ORF1b encodes from nsp1 to nsp16 proteins.
3. Mutations in ORF1a, ORF1b, and RdRp
ORF1a and ORF1b encode replicase polyproteins essential component of the viral RNA replication. Therefore, conserved structure of them have been the focus of antiviral drugs [18]. RNA-dependent RNA polymerase (RdRp) located between ORF1a and ORF1b is an important component for application and translation and has a functionally conserved region known as high sequence similarity [19]. The conserved region of RdRp has been widely used as a target for designing primers and probes based on RT-PCR technology in genetic diagnosis of coronavirus disease 2019 (COVID-19). In this regard, the mutations from ORF1a and ORF1b were analyzed, and we identified 302 types of synonymous and 530 types of non-synonymous mutation in ORF1a, and 179 types of synonymous and 305 types of non-synonymous mutation in ORF1b. Also, a total of 144 types of mutation were identified in the RdRp region corresponding to the 13,442 to 16,236 genomic position, of which 56 types of synonymous and 88 types of non-synonymous mutation were detected (Figure 6). Since the non-synonymous mutation with the highest frequency of in the RdRp region was P214L which determines the G.1 subclade together with G204R and R203K of the N gene, it was necessary to further study the functional interaction amongst these mutations of the ORF1a and N gene. On one hand, 9 types of primer-template mismatch were identified in the RdRp region. Although it is not understood how these mismatches affect the stability of primer-template complexes, it is necessary to determine the interrelationships.
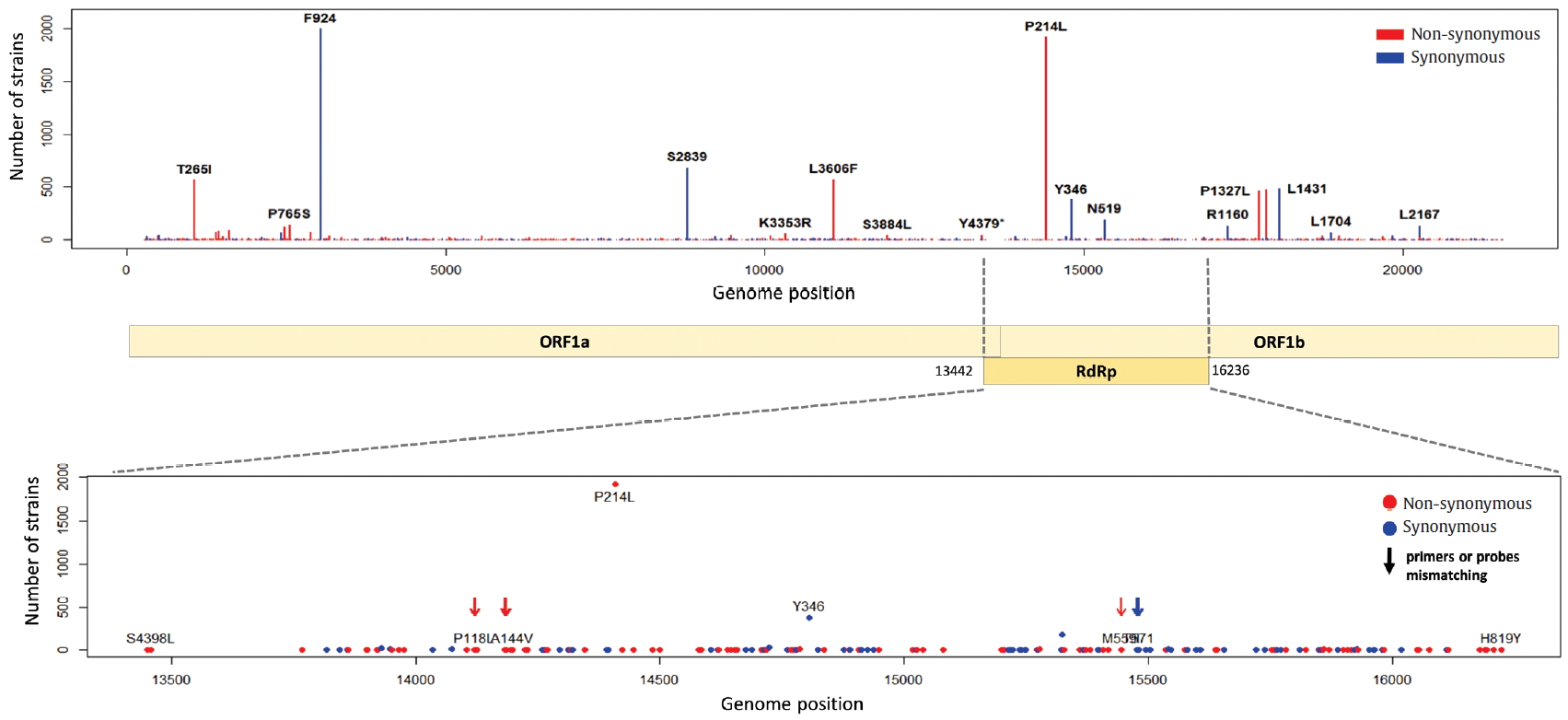
Distribution of mutations and primer-template mismatches in ORF1a and ORF1b. The genomic positions (13,442-16,236) of RNA-dependent RNA polymerase (RdRp) was based on NCBI ID NC_045512.2. The primer- and probe-template regions were derived from the lists published on the Centers for Disease Control and Prevention (CDC).
4. Mutations in S gene, receptor-binding domain and polybasic cleavage site
The S gene of SARS-CoV-2 plays an important role in escaping the immune system of host. The RBD in the S gene is directly or indirectly concerned with the binding and affinity to ACE2. The strong binding between RBD and ACE2 may have led to the worldwide transmission and rapid infectivity of SARS-CoV-2. On the other hand, the RRAR, which is located between S1 and S2 (681-685 amino acid position of the S gene), may be related to the pathogenicity of SARS-CoV-2. Therefore, it is might be inferred from the mutations in the RBD and the polybasic cleavage site in the S gene that the mutations will provide important clues to understanding immunity and pathogenicity of SARS-CoV-2. We identified 109 types of synonymous and 182 types of non-synonymous mutation in the S gene (Figure 7). D614G is a major non-synonymous mutation in G clade and accounts for 1,764 (13.0%) in a total of 13,537 non-synonymous mutations. Above all, it is located in the S1-S2 junction region near the polybasic cleavage site, but its biochemical and structural relationships with ACE2 has been unclear so far. In the RBD region (22,478-23,191 genomic position), we found 12 types of synonymous and 27 types of non-synonymous mutation. Among them, the D467V, I468F, I468T, I472V, G476S, S477G, V483A, P491R, Y508H, R509K, and V510L were located within the zone (443-510 amino acid position of the S gene) adjacent to ACE2. In particular, the V483A and G476S mutations have previously been reported to be related to human receptor-binding affinity in MERS and SARS-CoV research [20, 21]. We detected a R682Q mutation from the RRAR, where arginine residue was replaced by glutamine residue. The arginine residue is not only electrically charged but also strongly basic, while the glutamine residue is polar uncharged. Therefore, it is necessary to study how this biochemical difference may affect functional and structural changes of the proteins of S1 or S2. According to a recent study, the fact that a deletion of the QTQTN amino acid motif near the polybasic cleavage site is related to adaptation of SARS-CoV-2 has been reported. Hence, we tried to confirm whether some deletions occurred in this region (675-679 amino acid position of the S gene). We identified no deletion of QTQTN in this region, but confirmed 5 types of non-synonymous mutation (Type 1 with N679Y and S680P, Type 2 with N679K, Type 3 with Q677H, Type 4 with Q675H, and Type 5 with Q675R). Except for Type 1, Type 2 to 5 had a common feature where glutamine and asparagine with a polar uncharged side chain were converted into lysine, histidine, or arginine with basic property. Although deletion of the QTQTN motif was not identified near the polybasic cleavage site, it may be informative to study the functional significance of the five mutations in this region.
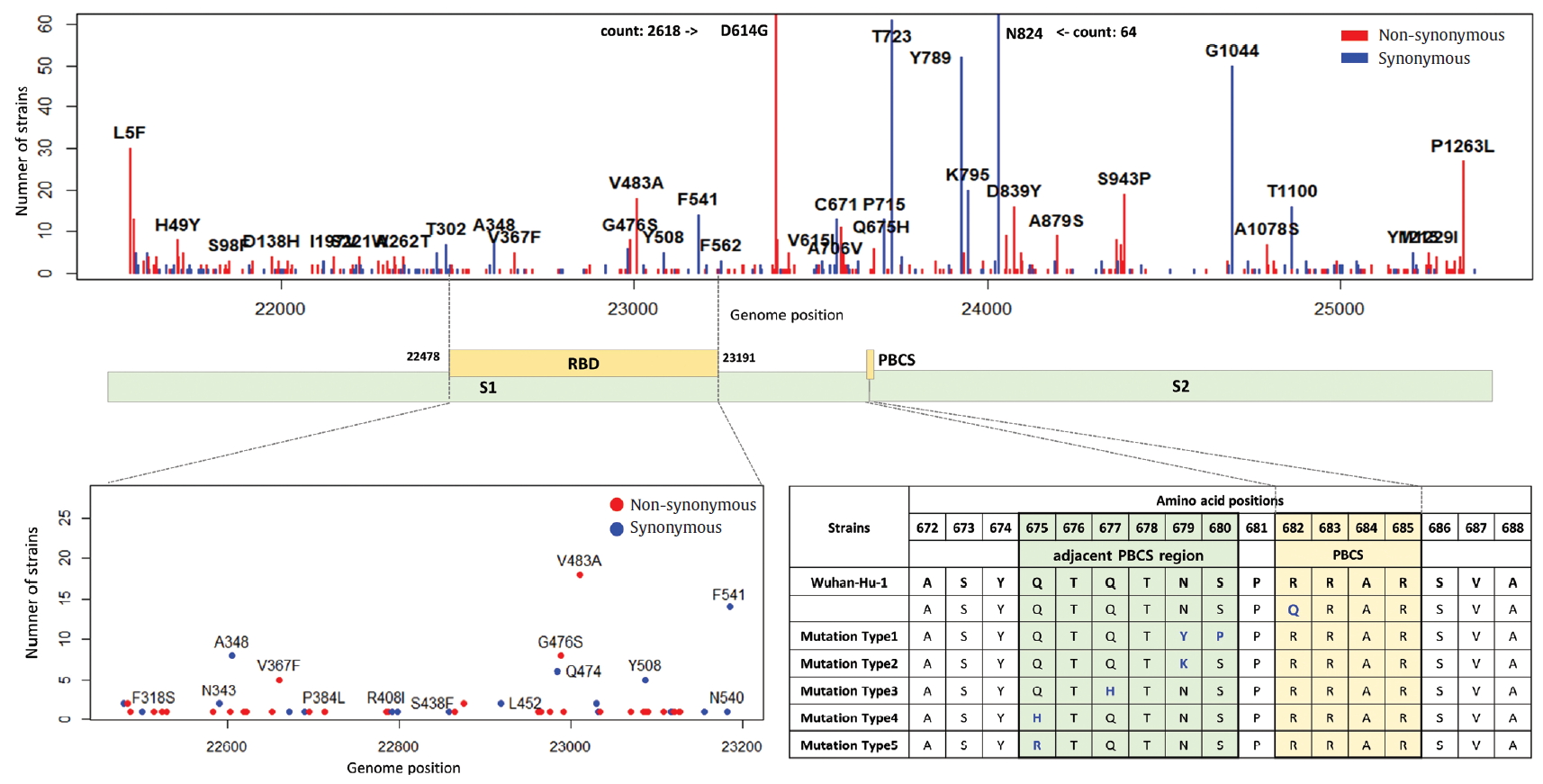
Distribution of mutations in receptor-binding domain and polybasic cleavage site within S gene. The genomic positions (22,478-23,191) of receptor-binding domain (RBD) and the amino acid position (682-685) of polybasic cleavage site (PBCS) were based on the reference [4], and the reference [14], respectivity.
5. Mutations in E, M, N and open reading frames encoding accessory proteins
The E, M, ORF6, ORF7a, ORF7b, ORF8, and ORF10 are less frequent mutations than other genes. ORF3a is a novel short positive protein essential to viral adaptation in vitro and is involved in the viral pathogenicity. The major types of synonymous mutation of ORF3a are Q57H and G251V, which are significant mutations in determining G.2 clade, and V clade, respectively. It is inferred from this result that Q57H and G251V may have a positive effect on viral adaptation. In the E gene known to be involved in viral budding along with the M gene, we confirmed a relatively small number of mutations in it. This means that there are high conserved regions, so it seems reasonable to infer that the preserved sequence is important to encode protein participating in viral reproduction. Like the region of RdRp, the E gene has been also used to design primers and probes for the diagnosis of SARS-CoV-2. We identified 10 types of synonymous and 12 types of non-synonymous mutation in the E gene (Figure 8). Of these mutations, 7 primer-template mismatch mutations (3 types of synonymous and 4 types of non-synonymous mutation) occurred. The M gene which is associated with cellular immunogenicity, showed 28 types of synonymous and 21 types of non-synonymous mutation, of which mutations with high frequency were observed as T175M and D3G. Several types of synonymous mutation in ORF6, ORF7, and ORF10 were identified, but mutations with high frequency were not observed. However, L84S in ORF8 was identified as a mutation with high frequency. The N gene packaging of the viral RNA genome into the medical ribonucleocapsid has been regarded as a candidate target region for primers and probes like the E and RdRp genes. In the N gene, we detected 63 types of synonymous and 119 types of non-synonymous mutation. Among them, R203K and G204R showed high frequencies in G.2 clade, together with P214L (ORF1b). In addition, 51 primer-template mismatch mutations (17 types of synonymous and 34 types of non-synonymous mutation) occurred.
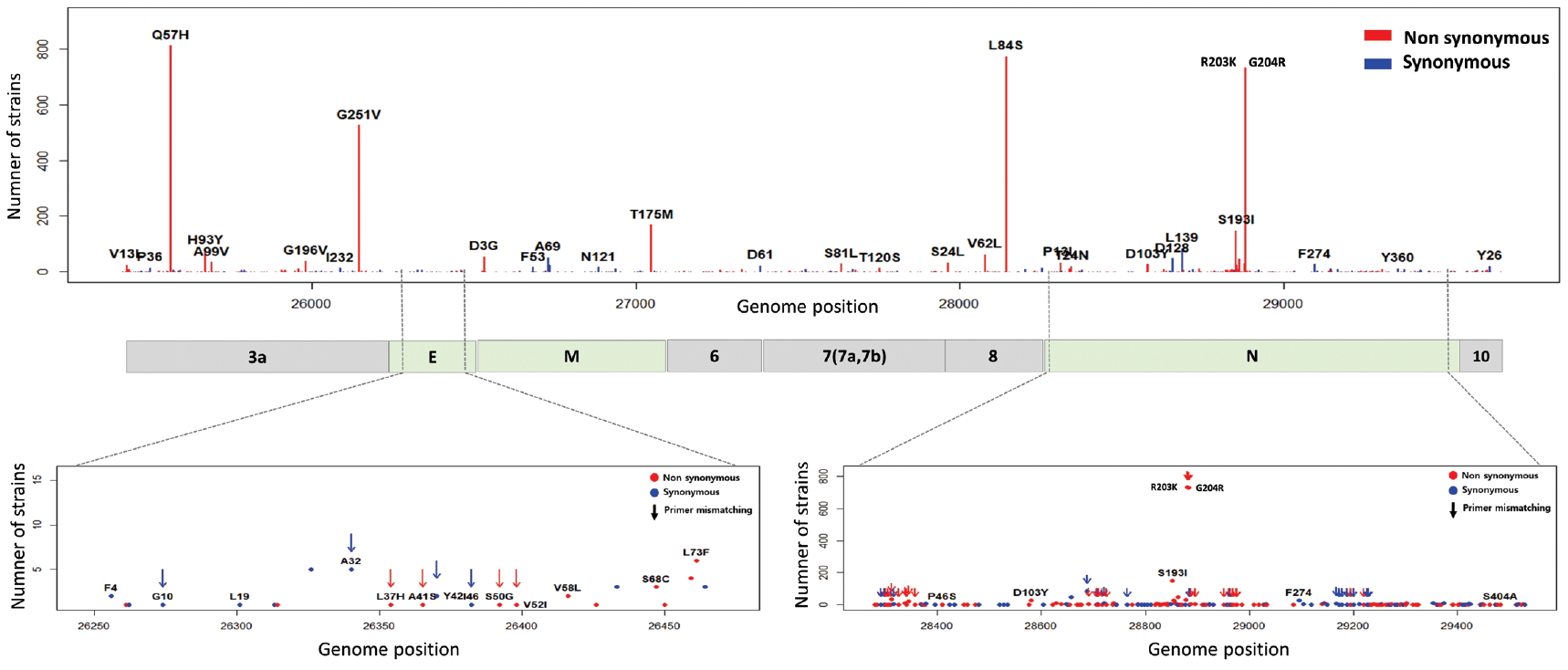
Distribution of mutations and primer-template mismatches in ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N, and ORF10. The genomic positions (13,442-16,236) of RNA-dependent RNA polymerase (RdRp) was based on NCBI ID NC_045512.2. The primer- and probe-template regions were retrieved from the lists published on the Centers for Disease Control and Prevention (CDC).
Discussion
From the non-synonymous mutation is overall more frequent than the synonymous mutations in the set of 4,254 SARS-CoV-2 genome sequences, it is inferred from this results that the evolution of the SARS-CoV-2 has been accepted to be positive selection. In consequence, several point mutations may directly or indirectly influence the interaction between SARS-CoV-2 and human, and the diversity of mutations in SARS-CoV-2 may enhance the evolution of the virus towards rapid transmission. Based on our analysis, the D614G (S) mutation of G clade, which revealed the highest frequent in our phylogenetic analysis, may have a positive advantage in natural selection. According to the frequency of mutations within SARS-CoV-2 strains, ORF1a, ORF1b, S, and N genes were shown at high frequency, and they may advantageously evolve to adapt to not only external interactions with host cells (such as recognizing a cell surface receptor, attaching to the host receptor, and fusing with cellular membranes), but also internal interactions in host cells (such as replicating and transcribing viral genome, and budding by cellular exocytosis). Although D614G in the S gene is not a mutation within RBD, the fact that it occurred as the highest frequency in all SARS-CoV-2 genomes suggests it may be related to host infection and transmission. Therefore, we believe that it is necessary to continuously monitor the accumulation of mutations and to further study how these mutations affect receptor affinity, propagation ability, and pathogenicity. On the other hand, RdRp, E, and N genes are the target genes for designing primers and probes in RT-PCR-based SARS-CoV-2 diagnosis, owing to their high sequence conservation. It has not been known how the primer-template mismatches affect the accuracy and precision of the genetic diagnosis of COVID-19, but we suggest that it is desirable to avoid the variable hotspot regions as much as possible. Our results about genomic mutations of SARS-CoV-2 strains may be helpful for interpreting the potential relationships of pathogenicity, infectivity, and transmission between SARS-CoV-2 and human host.
Acknowledgements
We acknowledge the incessant devotion and actively hard work of the staffs in the Division of Viral Diseases Research from the Korea Centers for Disease Control and Prevention. This study was funded by Korea Centers for Disease Control and Prevention (no.: 4800-4837-301).
Notes
Conflicts of Interest
The authors have no conflicts of interest to declare.