Articles
- Page Path
- HOME > Osong Public Health Res Perspect > Volume 12(1); 2021 > Article
-
Brief Report
Genomic Surveillance of SARS-CoV-2: Distribution of Clades in the Republic of Korea in 2020 - Ae Kyung Parka, Il-Hwan Kima, Junyoung Kimb, Jeong-Min Kima, Heui Man Kima, Chae young Leea, Myung-Guk Hanc, Gi-Eun Rhied, Donghyok Kwone, Jeong-Gu Namf, Young-Joon Parkg, Jin Gwackg, Nam-Joo Leea, SangHee Wooa, Jin Sun Nod, Jaehee Leea, Jeemin Hah, JeeEun Rheea, Cheon-Kwon Yooi, Eun-Jin Kima
-
Osong Public Health and Research Perspectives 2021;12(1):37-43.
DOI: https://doi.org/10.24171/j.phrp.2021.12.1.06
Published online: February 23, 2021
aDivision of Emerging Infectious Diseases, Bureau of Infectious Diseases Diagnosis Control, Korea Disease Control and Prevention Agency, Cheongju, Korea
bDivision of Bacterial Diseases, Bureau of Infectious Diseases Diagnosis Control, Korea Disease Control and Prevention Agency, Cheongju, Korea
cDivision of Viral Diseases, Bureau of Infectious Diseases Diagnosis Control, Korea Disease Control and Prevention Agency, Cheongju, Korea
dDivision of High-Risk Pathogens, Bureau of Infectious Diseases Diagnosis Control, Korea Disease Control and Prevention Agency, Cheongju, Korea
eDivision of Public Health Emergency Response Research, Bureau of Public Health Emergency Preparedness, Korea Disease Control and Prevention Agency, Cheongju, Korea
fDivision of Laboratory Diagnosis Analysis, Capital Regional Center for Disease Control and Prevention, Seoul, Korea
gCentral Disease Control Headquarters Epidemiological Investigation Team, Korea Disease Control and Prevention Agency, Cheongju, Korea
hDivision of Laboratory Diagnosis Analysis, Chungcheong Regional Center for Disease Control and Prevention, Daejeon, Korea
iBureau of Infectious Diseases Diagnosis Control, Korea Disease Control and Prevention Agency, Cheongju, Korea
- *Corresponding author: Eun-Jin Kim, Division of Emerging Infectious Diseases, Bureau of Infectious Diseases Diagnosis Control, Korea Disease Control and Prevention Agency, Cheongju, Korea, E-mail: ekim@korea.kr
- † These authors contributed equally to this work.
©2021 Korea Disease Control and Prevention Agency
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Abstract
- Since a novel beta-coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first reported in December 2019, there has been a rapid global spread of the virus. Genomic surveillance was conducted on samples isolated from infected individuals to monitor the spread of genetic variants of SARS-CoV-2 in Korea. The Korea Disease Control and Prevention Agency performed whole genome sequencing of SARS-CoV-2 in Korea for 1 year (January 2020 to January 2021). A total of 2,488 SARS-CoV-2 cases were sequenced (including 648 cases from abroad). Initially, the prevalent clades of SARS-CoV-2 were the S and V clades, however, by March 2020, GH clade was the most dominant. Only international travelers were identified as having G or GR clades, and since the first variant 501Y.V1 was identified (from a traveler from the United Kingdom on December 22nd, 2020), a total of 27 variants of 501Y.V1, 501Y.V2, and 484K.V2 have been classified (as of January 25th, 2021). The results in this study indicated that quarantining of travelers entering Korea successfully prevented dissemination of the SARS-CoV-2 variants in Korea.
- Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first reported in December 2019, in Wuhan, China, and subsequently, the virus has spread rapidly all over the world [1]. Since the first novel coronavirus genome sequence was made available (January 10th, 2020 [2]), whole genome sequencing (WGS) has been extensively performed globally, to characterize the virus. This has led to many important findings, and more than 412,699 complete genome sequences have been made available in a global initiative on sharing all influenza data (GISAID [as of January 25th, 2021]) [3]. Detection of novel variants of SARS-CoV-2 including 501Y.V1 (also known as Variant of Concern 2020/12/01) [4], 501Y.V2 [5] and 484K.V2 [6], which have rapidly spread worldwide, have emphasized the need for sequence-based strain surveillance, to promptly detect mutations to prevent the spread of new variants.
- The Republic of Korea was one of the first countries to report a significant Coronavirus Disease-19 (COVID-19) outbreak which was managed successfully due to the effective cooperation between the government and the people of Korea. As part of a comprehensive national surveillance of COVID-19, the Korea Disease Control and Prevention Agency (KDCA) conducted the WGS of SARS-CoV-2 using real-time genomic epidemiology data to monitor how the virus mutates, and correlates with sequence variation since the first case of SARS-CoV-2 infection was identified in Korea on January 20th, 2020 [7].
- In this Brief Report, the SARS-CoV-2 genomes of 2,488 confirmed cases in Korea were sequenced and analyzed, in the context of the global viral population, from the beginning of the pandemic to provide valuable insights into the genetic pattern of SARS-CoV-2 distribution in Korea.
Introduction
- Nasopharyngeal and oropharyngeal swabs were collected from SARS-CoV-2, real-time reverse transcription (RT)-PCR confirmed cases. RNA extraction and RT-PCR were performed on the samples from the swabs, according to the methods described in a previous report [8].
- To perform WGS, cDNA was amplified using primer pools (https://artic.network/ncov-2019). Libraries were prepared using the Nextera DNA Flex Library Prep Kit (Illumina, USA), and sequenced on the MiSeq instrument (Illumina, USA) with 2 × 150 base pairs using a MiSeq reagent kit V2 (Illumina, USA) to obtain an average genome coverage greater than 1,000 × for all the isolates. The sequence reads were trimmed and mapped to reference genome MN908947.3 using CLC Genomics Workbench Version 20.0.3 (CLC Bio, Denmark).
- All types of clades (including variants) were isolated by the KDCA (Division of Emerging Infectious Diseases) using the Vero E6 cell line and were deposited in the National Culture Collection for Pathogens and sequence data were uploaded to GISAID (Table 1).
- The study was approved by the International Review Board at the Korea Disease Control and Prevention Agency (2020-03-01-P-A). The board waived the requirement for written consent.
Materials and Methods
- 1. SARS-CoV-2 genomic surveillance in Korea
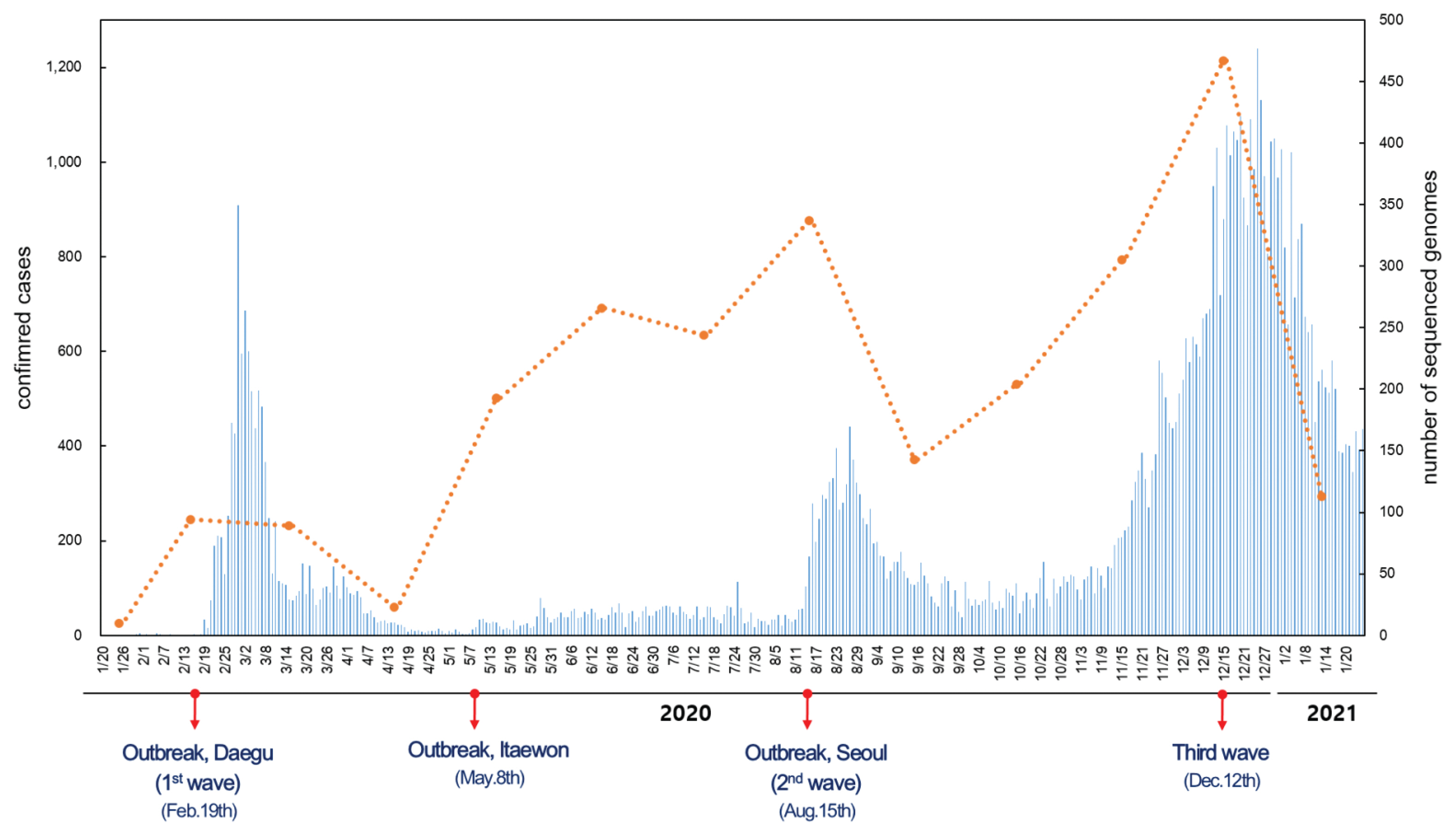
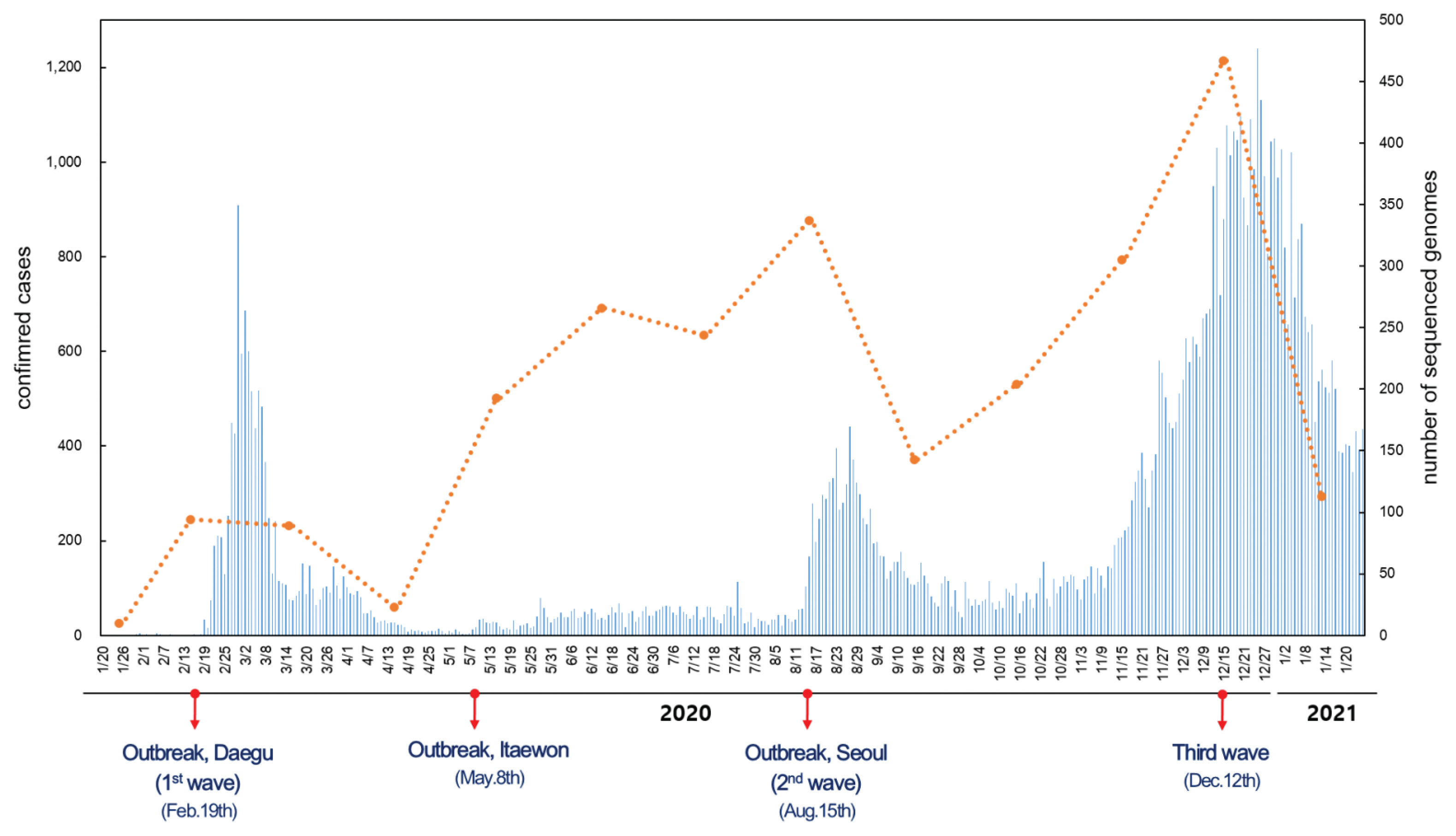
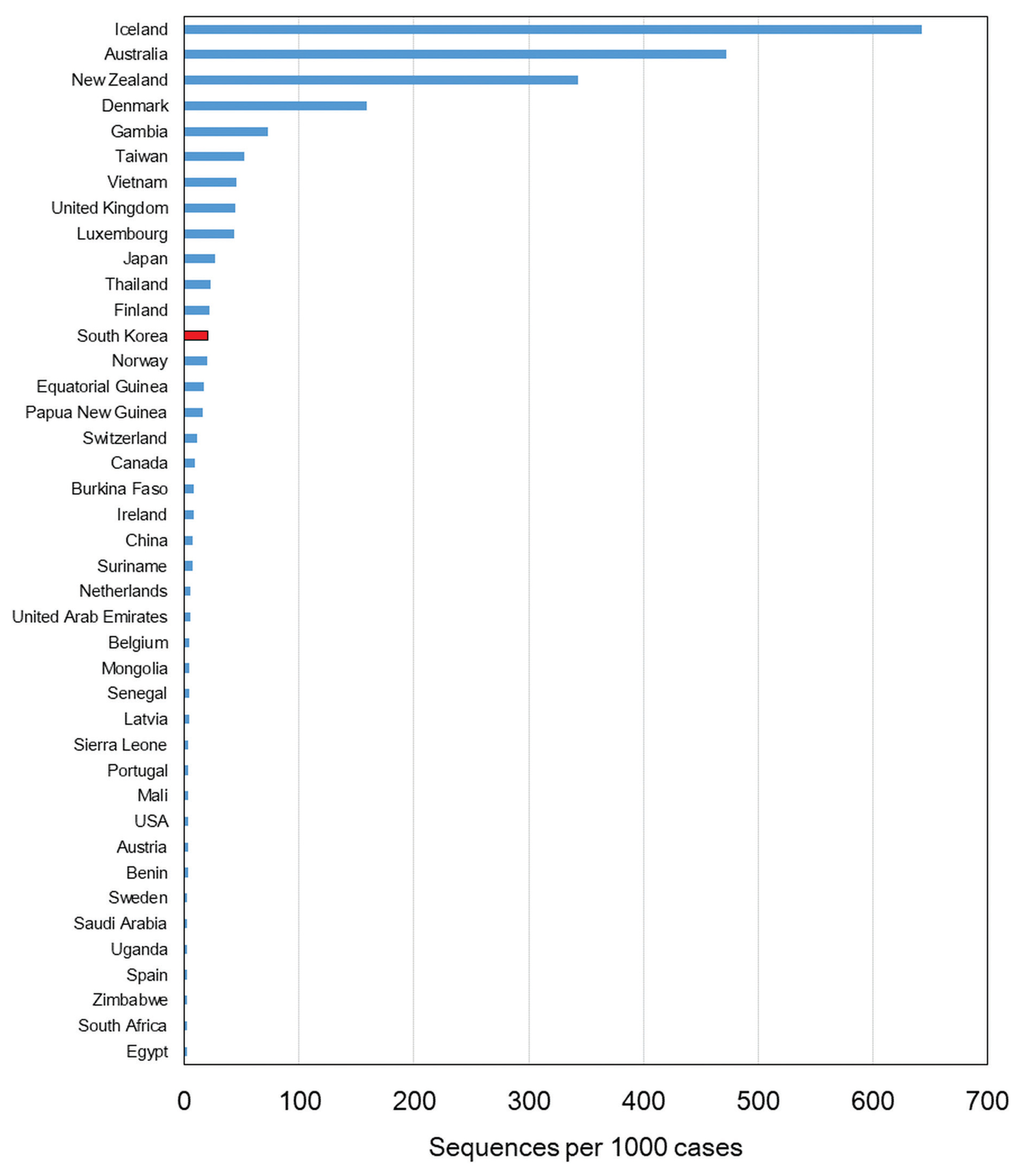
- Since the first confirmed case reported on January 20th, 2020, the number of confirmed cases has reached 75,521 as of January 25th, 2021. The number of cases imported from abroad were 6,144. The daily number of reported COVID-19 cases in Korea is shown in Figure 1. The first wave of COVID-19, which was associated with the Shincheonji outbreak, began in February and lasted until April 2020 [9]. In May 2020, another outbreak of COVID-19 was reported which was traced to the Itaewon district in Seoul. A second wave of COVID-19 in Korea began in August 2020, and a third wave in November 2020, both originating in the Seoul area. As indicated by the number of sequenced genomes, WGS in Korea has covered more than 27% of the total number of confirmed cases as of May 2020. There was a remarkable increase in confirmed cases during the third wave which resulted in a decrease of sequencing during this period, but overall, the sequencing of SARS-CoV-2 in Korea was about 3.3% of all COVID-19 confirmed cases. Based on the percentage of COVID-19 cases which were genome sequenced, and uploaded to GISAID, Korea ranked in the top 15 countries (Figure 2).
- 2. Distribution of clades in Korea
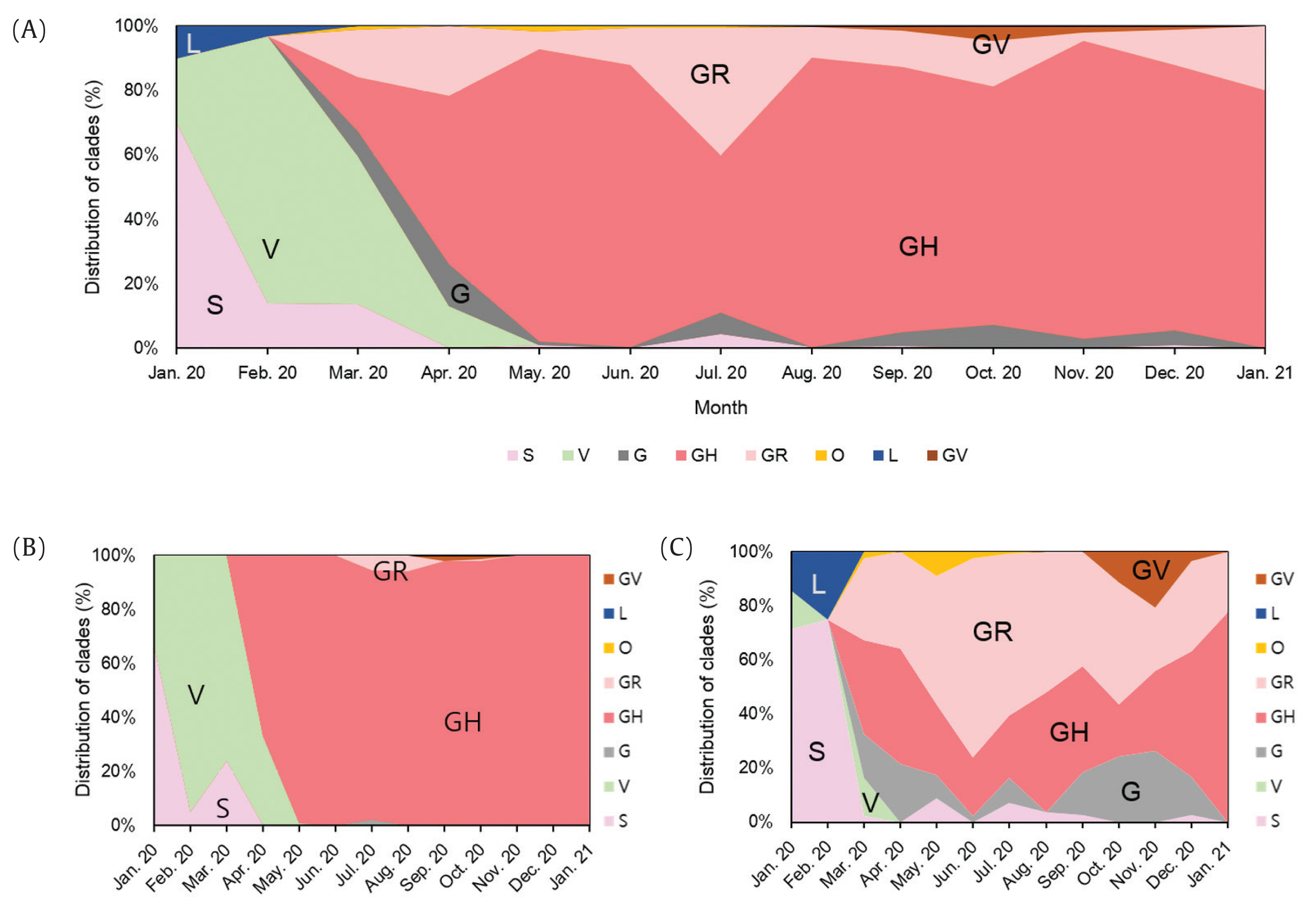
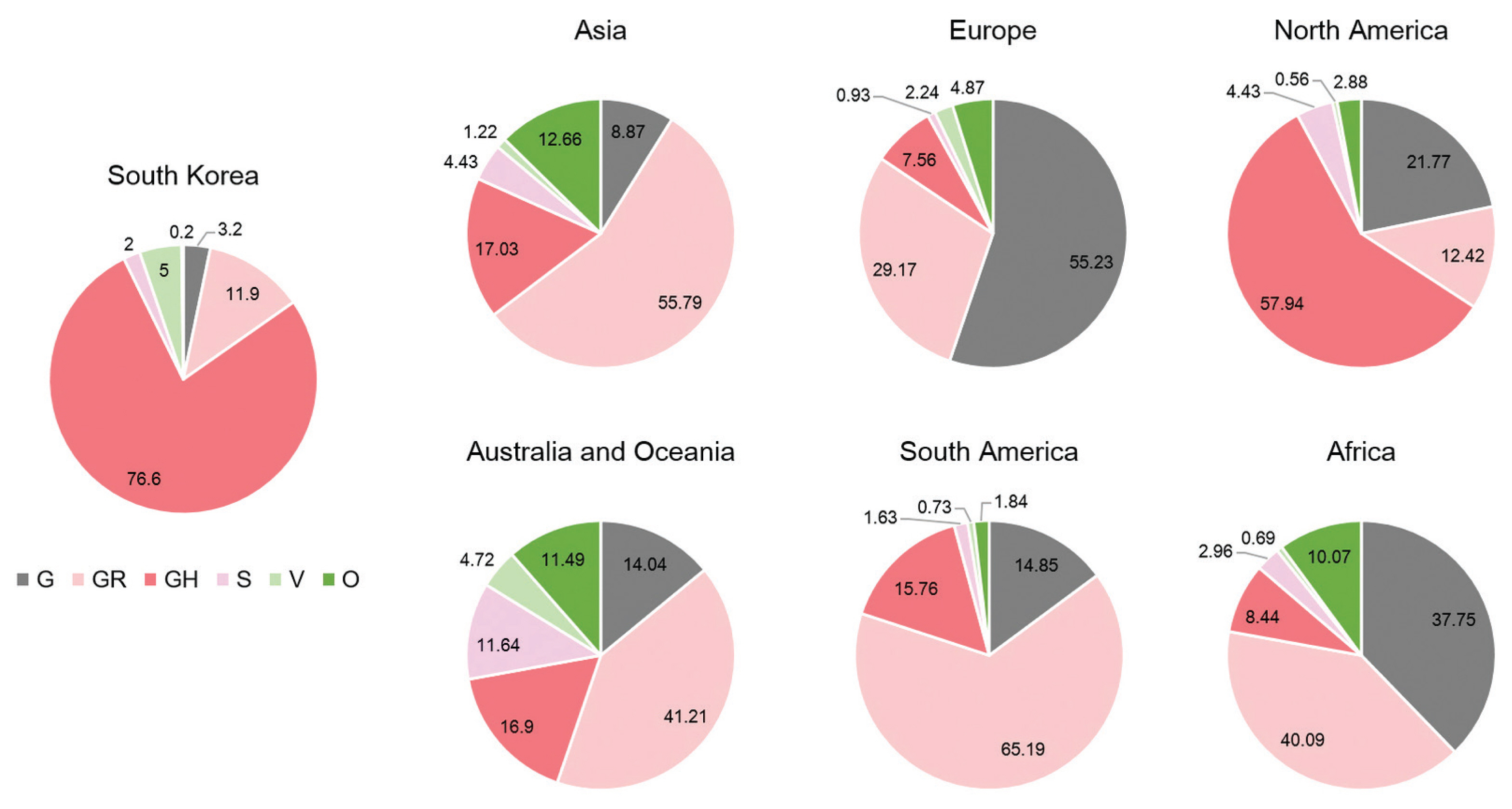
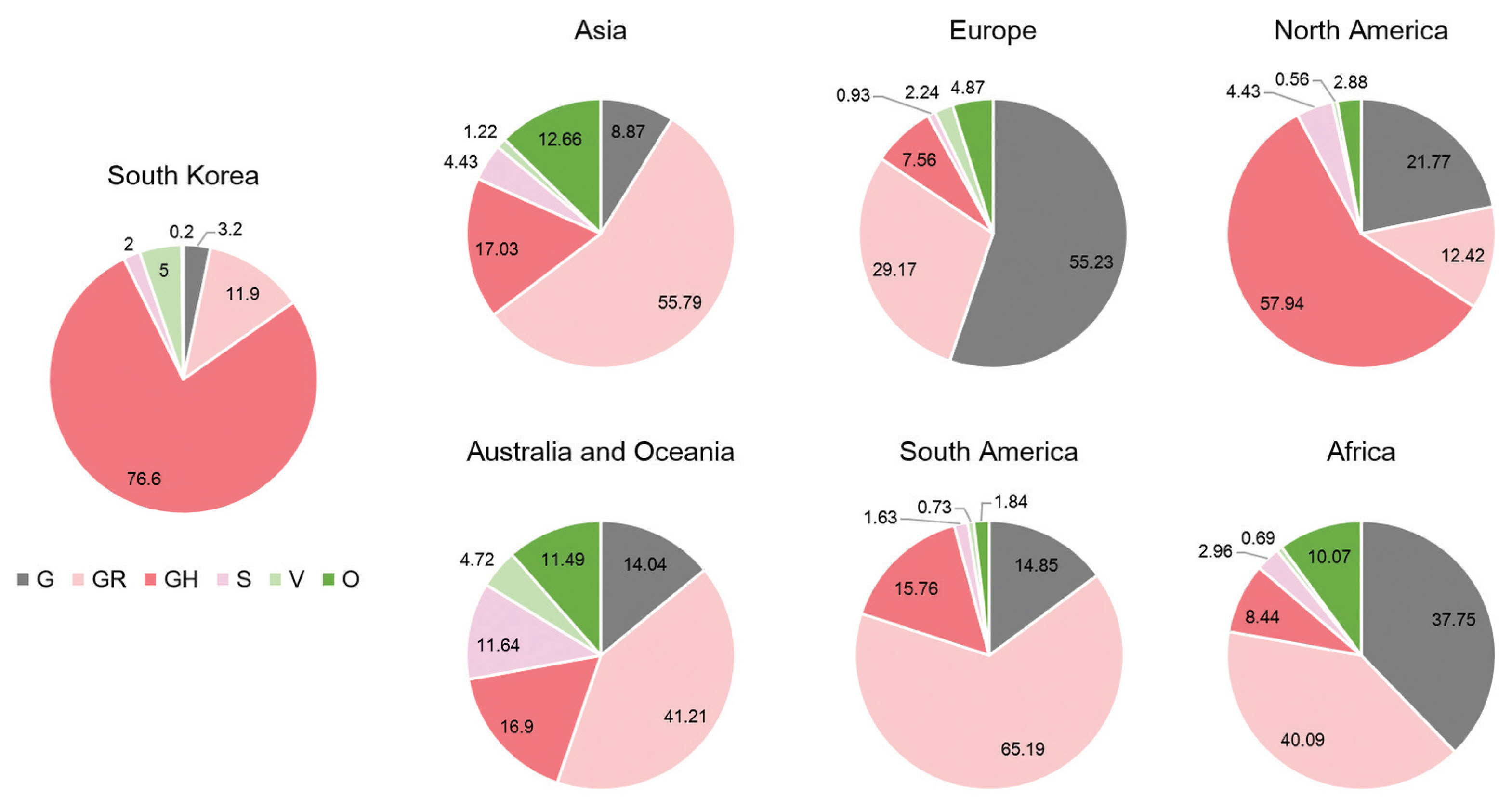
- A total of 2,488 SARS-CoV-2 were sequenced during the period between January 20th, 2020 and January 25th (2,021 including 648 cases imported from abroad). The overall distribution of clades over time is shown in Figure 3A. There was an initial period during January to March 2020 when the S and V clades (GISAID nomenclatures) were more prevalent in total than G/GR/GH clades in Korea (Figure 3B). S and V clades were the most prevalent clades at the beginning of the epidemic however, after the emergence of the variant D614G spike protein mutation, this variant became predominant [10]. The G/GR/GH clades were first identified in March 2020 in Korea, with the GH clade becoming dominant. Interestingly, the G/GR clades identified in Korea, were only isolated from international travelers, and the GR clades were only isolated from cases in close contact with someone who came from abroad indicating there was a low prevalence of GR in Korea. The diversity of clades observed in cases imported from abroad compared with domestic cases, led to the belief that COVID-19 from abroad had been restricted through comprehensive, and strict quarantine and disease control measures (Figure 3B). A comparison of the prevalent clades in each continent highlights a higher prevalence of the GH clade in Korea (Figure 4).
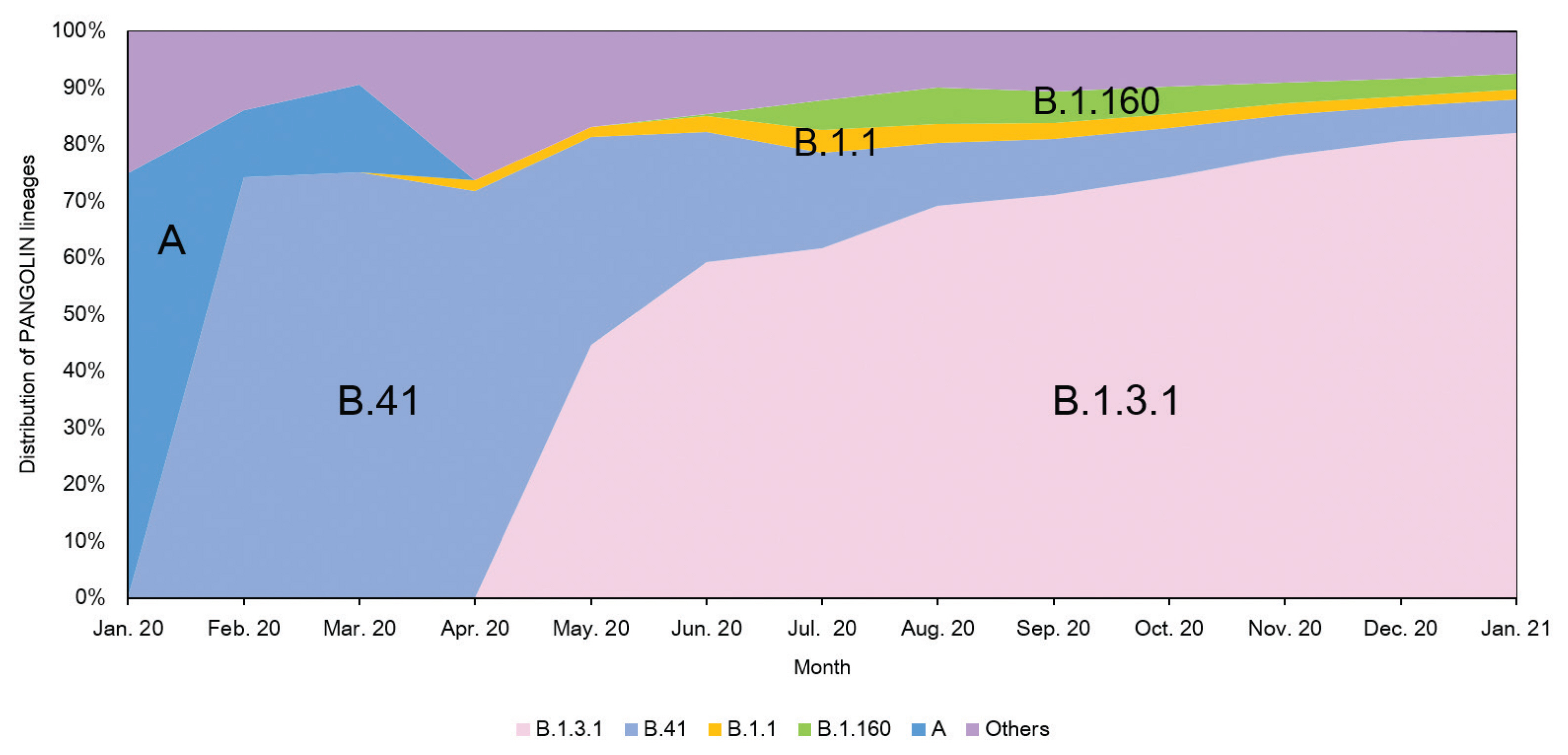
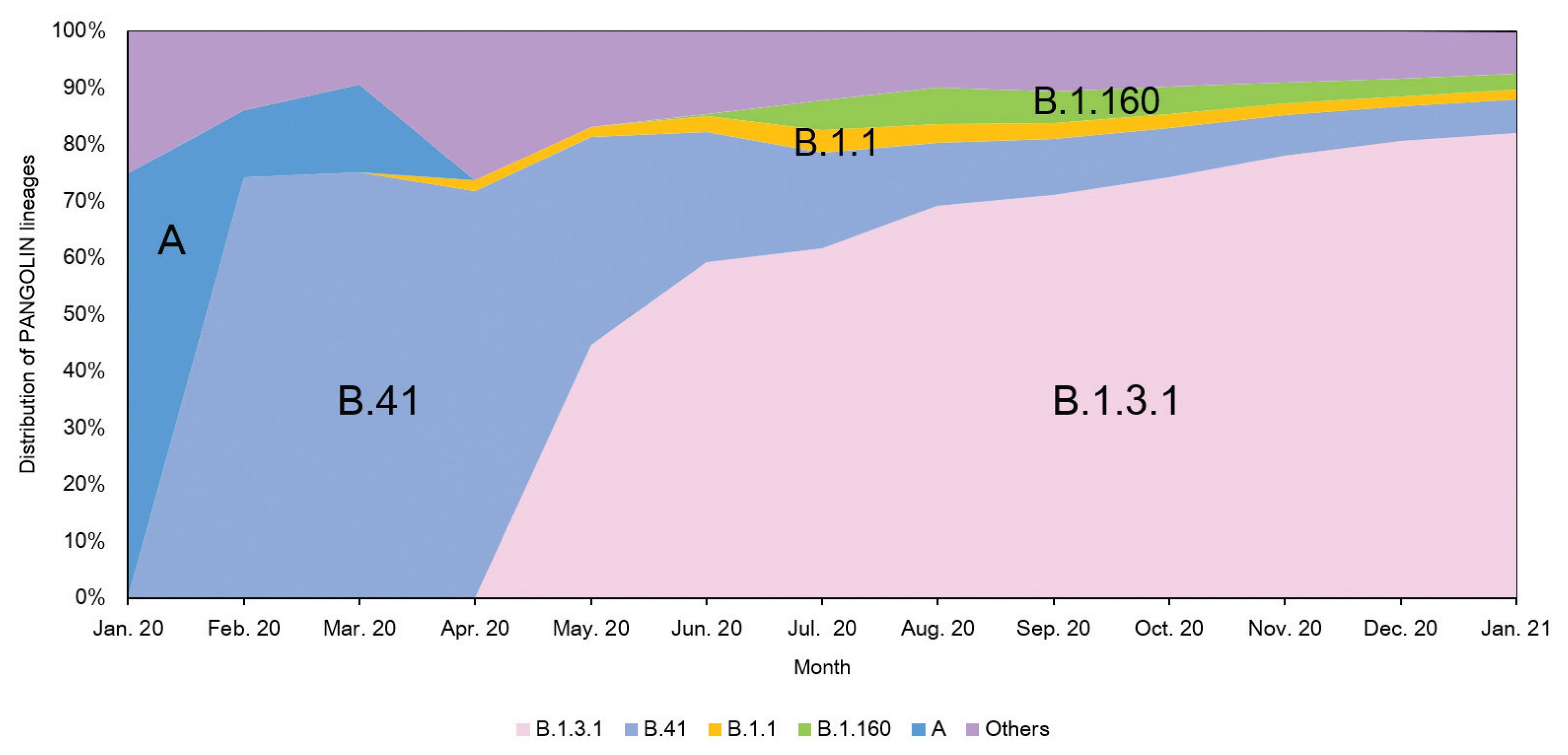
- The analysis of domestic cases of SARS-CoV-2 in Korea using the Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN v2.1.7) database [11] indicated that over 80% of isolates were classified as B.1.3.1 (as of January 25th, 2021). The proportion of the B.1.3.1 lineage has continued to rise steadily as shown in Figure 5. Analysis of domestic cases also supports that SARS-CoV-2 imported from abroad does not seem to be spreading within Korea because the lineage distributions was the same (B.1.3.1) from May 2020 to January 2021.
- 3. Multiple SARS-CoV-2 variants
- Multiple SARS-CoV-2 variants have been circulating globally. The main variants include 501Y.V1 (B.1.1.7), 501Y.V2 (B.1.351), and 484K.V2 (P.1 and P.2). The nomenclatures in parentheses are assigned by the PANGOLIN database which was developed by members of the COVID-19 Genomics UK team [11]. The PANGOLIN database was deployed to establish the transmission patterns of numerous variants of the virus. The KDCA has carried out investigations into travelers who returned from abroad. The first 501Y.V1 variant was identified as originating from a person traveling from the United Kingdom on December 22nd, 2020, and an additional 18 cases were identified as of January 25th, 2021 (Table 1).
- The first 501Y.V2 variant was identified from a person traveling from South Africa on December 26th, 2020, and 4 additional cases were identified as of January 25th, 2021 (Table 1). The first 484K.V2 was identified from a person traveling from Brazil on January 10th, 2021, and 2 additional cases were confirmed as of January 25th, 2021 (Table 1).
- Among 19 cases of 501Y.V1, 2 were identified from people traveling from the Maldives and Ghana, on January 25th, 2021. Among 5 cases of 501Y.V2, 3 were confirmed from people traveling from Zimbabwe, Malawi, and Tanzania on January 25th, 2021. Through surveillance, 501Y.V1 was first identified from a person traveling from the Maldives, and 501Y.V2 from two people traveling from Zimbabwe and Malawi.
Results
- In January 2020, following the first case of COVID-19 in Korea, real-time genomic surveillance was being conducted by the KDCA. Based on the percentage of COVID-19 cases which were genome sequenced, and uploaded to GISAID (1,632 as of January 25th, 2021), Korea ranked in the top 15 countries.
- Analysis of genome sequences indicated that from January to March 2020, S and V were the prevalent clades in Korea. However, the prevalent clade of Korea is now GH (first isolated in March 2020). The other clades which are prevalent in Europe, G and GR, have not been identified in domestic cases of COVID-19. In addition, analysis of the PANGOLIN database lineages indicated that the majority of SARS-CoV-2 in Korea were grouped into the B.1.3.1 lineage. Therefore, these results indicated that quarantine for all travelers entering Korea played a crucial role in preventing the variants of SARS-CoV-2 spreading in Korea.
- Furthermore, genomic surveillance has also played a crucial role in monitoring recent variants including 501Y.V1, 501Y.V2, and 484K.V2. To date these variants of SARS-CoV-2 have not been introduced and spread in Korea, indicating that the quarantine and containment strategy was effective.
- Despite several sharp spikes in the number of confirmed COVID-19 cases in Korea in 2020, the cooperation between the Korean government and the general population has enabled a successful 3T strategy (Test, Trace, and Treat) for containing the COVID-19 pandemic. The KDCA will continue to contribute to a global effort to better understand, treat, and prevent COVID-19.
Discussion
-
Acknowledgements
- We are grateful to all the contributions from the laboratories responsible for obtaining the specimens and all the laboratories where genetic sequence data were generated, and shared via the GISAID, on which this research is based. Special thanks to the Emergency Operation Center of the KDCA for its great effort in recording new confirmed cases.
- This study was funded by Korea Disease Control and Prevention Agency (no.: 4800-4837-301, 4800-4834-303).
Acknowledgements

| SARS-CoV-2 clade | NCCP number | Collection date | GISAID accession number |
|---|---|---|---|
| S | 43326 | Jan 25, 2020 | EPI_ISL_407193 |
| L | 43330 | Feb 1, 2020 | EPI_ISL_412872 |
| V | 43342 | Feb 17, 2020 | EPI_ISL_497961 |
| G | 43344 | Mar 28, 2020 | EPI_ISL_506983 |
| GR | 43343 | Mar 27, 2020 | EPI_ISL_506981 |
| GH | 43345 | May 8, 2020 | EPI_ISL_510563 |
| GV | 43346 | Sep 29, 2020 | EPI_ISL_747316 |
| 501Y.V1* | 43381 | Dec 22, 2020 | EPI_ISL_738139 |
| 501Y.V2* | 43382 | Dec 26, 2020 | EPI_ISL_762992 |
| 484K.V2* | In progress | Jan 10, 2021 | EPI_ISL_833249 |
- 1. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579(7798). 270−3. PMID: 10.1038/s41586-020-2012-7. PMID: 32015507. PMID: 7095418.PubMedPMC
- 2. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020;579(7798). 265−9. PMID: 10.1038/s41586-020-2008-3. PMID: 32015508. PMID: 7094943.ArticlePubMedPMC
- 3. Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill 2017;22(13). 30494PMID: 10.2807/1560-7917.ES.2017.22.13.30494. PMID: 28382917. PMID: 5388101.ArticlePubMedPMC
- 4. Rambaut A, Loman N, Pybus O, et al. [Internet]. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Viological.org 2020 Available from: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563.
- 5. Tegally H, Wilkinson E, Giovanetti M, et al. [Preprint]. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv: 2020.12.21.20248640v1. 2020 Available from: https://www.medrxiv.org/content/10.1101/2020.12.21.20248640v1.Article
- 6. Faria NR, Claro IM, Candido D, et al. [Internet]. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus Preliminary findings. Viological.org 2020 Available from: https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586.
- 7. Kim JY, Choe PG, Oh Y, et al. The First Case of 2019 Novel Coronavirus Pneumonia Imported into Korea from Wuhan, China: implication for infection prevention and control measures. J Korean Med Sci 2020;35(5). e61PMID: 10.3346/jkms.2020.35.e61. PMID: 32030925. PMID: 7008073.ArticlePubMedPMC
- 8. Kim JM, Chung YS, Jo HJ, et al. Identification of Coronavirus Isolated from a Patient in Korea with COVID-19. Osong Public Health Res Perspect 2020;11(1). 3−7. PMID: 10.24171/j.phrp.2020.11.1.02. PMID: 32149036. PMID: 7045880.ArticlePubMedPMC
- 9. Kim J-M, Park SY, Lee D, et al. [Preprint]. Genomic investigation of the coronavirus disease-2019 outbreak in the Republic of Korea. Research Square: rs-65856 2020 Available from: https://www.researchsquare.com/article/rs-65856/v1.Article
- 10. Isabel S, Grana-Miraglia L, Gutierrez JM, et al. Evolutionary and structural analyses of SARS-CoV-2 D614G spike protein mutation now documented worldwide. Sci Rep 2020;10(1). 14031PMID: 10.1038/s41598-020-70827-z. PMID: 32820179. PMID: 7441380.ArticlePubMedPMC
- 11. Rambaut A, Holmes EC, Hill V, et al. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. Nat Microbiol 2020;5(11). 1403−7. PMID: 10.1038/s41564-020-0770-5. PMID: 32669681.ArticlePubMedPMC
- 12. Elbe S, Buckland-Merrett G. Data, disease, and diplomacy: GISAID’s innovative contribution to global health. Glob Chall 2017;1(1). 33−46. PMID: 10.1002/gch2.1018. PMID: 31565258. PMID: 6607375.ArticlePubMedPMC
- 13. Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis 2020;20(5). 533−4. PMID: 10.1016/S1473-3099(20)30120-1. PMID: 32087114. PMID: 7159018.ArticlePubMedPMC
References
Figure & Data
References
Citations
- Increased viral load in patients infected with severe acute respiratory syndrome coronavirus 2 Omicron variant in the Republic of Korea
Jeong-Min Kim, Dongju Kim, Nam-Joo Lee, Sang Hee Woo, Jaehee Lee, Hyeokjin Lee, Ae Kyung Park, Jeong-Ah Kim, Chae Young Lee, Il-Hwan Kim, Cheon Kwon Yoo, Eun-Jin Kim
Osong Public Health and Research Perspectives.2023; 14(4): 272. CrossRef - Rapid Emergence of the Omicron Variant of Severe Acute Respiratory Syndrome Coronavirus 2 in Korea
Ae Kyung Park, Il-Hwan Kim, Chae Young Lee, Jeong-Ah Kim, Hyeokjin Lee, Heui Man Kim, Nam-Joo Lee, SangHee Woo, Jaehee Lee, JeeEun Rhee, Cheon-Kwon Yoo, Eun-Jin Kim
Annals of Laboratory Medicine.2023; 43(2): 211. CrossRef - A Seroprevalence Study on Residents in a Senior Care Facility with Breakthrough SARS-CoV-2 Omicron Infection
Heui Man Kim, Eun Ju Lee, Sang Won O, Yong Jun Choi, Hyeokjin Lee, Sae Jin Oh, Jeong-Min Kim, Ae Kyung Park, Jeong-Ah Kim, Chae young Lee, Jong Mu Kim, Hanul Park, Young Joon Park, Jeong-Hee Yu, Eun-Young Kim, Hwa-Pyeong Ko, Eun-Jin Kim
Viral Immunology.2023; 36(3): 203. CrossRef - COVID-19 Cases and Deaths among Healthcare Personnel with the Progression of the Pandemic in Korea from March 2020 to February 2022
Yeonju Kim, Sung-Chan Yang, Jinhwa Jang, Shin Young Park, Seong Sun Kim, Chansoo Kim, Donghyok Kwon, Sang-Won Lee
Tropical Medicine and Infectious Disease.2023; 8(6): 308. CrossRef - The COVID-19 pandemic and healthcare utilization in Iran: evidence from an interrupted time series analysis
Monireh Mahmoodpour-Azari, Satar Rezaei, Nasim Badiee, Mohammad Hajizadeh, Ali Mohammadi, Ali Kazemi-Karyani, Shahin Soltani, Mehdi Khezeli
Osong Public Health and Research Perspectives.2023; 14(3): 180. CrossRef - Online Phylogenetics with matOptimize Produces Equivalent Trees and is Dramatically More Efficient for Large SARS-CoV-2 Phylogenies than de novo and Maximum-Likelihood Implementations
Alexander M Kramer, Bryan Thornlow, Cheng Ye, Nicola De Maio, Jakob McBroome, Angie S Hinrichs, Robert Lanfear, Yatish Turakhia, Russell Corbett-Detig, Olivier Gascuel
Systematic Biology.2023; 72(5): 1039. CrossRef - Genomic epidemiology of SARS-CoV-2 variants in South Korea between January 2020 and February 2023
Il-Hwan Kim, Jin Sun No, Jeong-Ah Kim, Ae Kyung Park, HyeokJin Lee, Jeong-Min Kim, Nam-Joo Lee, Chi-Kyeong Kim, Chae Young Lee, SangHee Woo, Jaehee Lee, JeeEun Rhee, Eun-Jin Kim
Virology.2023; 587: 109869. CrossRef - Genomic evidence of SARS‐CoV‐2 reinfection in the Republic of Korea
Ae Kyung Park, Jee Eun Rhee, Il‐Hwan Kim, Heui Man Kim, Hyeokjin Lee, Jeong‐Ah Kim, Chae Young Lee, Nam‐Joo Lee, SangHee Woo, Jaehee Lee, Jin Sun No, Gi‐Eun Rhie, Seong Jin Wang, Sang‐Eun Lee, Young Joon Park, Gemma Park, Jung Yeon Kim, Jin Gwack, Cheon‐K
Journal of Medical Virology.2022; 94(4): 1717. CrossRef - SARS-CoV-2 B.1.619 and B.1.620 Lineages, South Korea, 2021
Ae Kyung Park, Il-Hwan Kim, Heui Man Kim, Hyeokjin Lee, Nam-Joo Lee, Jeong-Ah Kim, SangHee Woo, Chae young Lee, Jaehee Lee, Sae Jin Oh, JeeEun Rhee, Cheon-Kwon Yoo, Eun-Jin Kim
Emerging Infectious Diseases.2022; 28(2): 415. CrossRef - Humoral and Cellular Responses to COVID-19 Vaccines in SARS-CoV-2 Infection-Naïve and -Recovered Korean Individuals
Ji-Young Hwang, Yunhwa Kim, Kyung-Min Lee, Eun-Jeong Jang, Chang-Hoon Woo, Chang-Ui Hong, Seok-Tae Choi, Sivilay Xayaheuang, Jong-Geol Jang, June-Hong Ahn, Hosun Park
Vaccines.2022; 10(2): 332. CrossRef - Increase in Viral Load in Patients With SARS-CoV-2 Delta Variant Infection in the Republic of Korea
Jeong-Min Kim, Jee Eun Rhee, Myeongsu Yoo, Heui Man Kim, Nam-Joo Lee, Sang Hee Woo, Hye-Jun Jo, Donghyok Kwon, Sangwon Lee, Cheon Kwon Yoo, Eun-Jin Kim
Frontiers in Microbiology.2022;[Epub] CrossRef - Molecular Dynamics Studies on the Structural Stability Prediction of SARS-CoV-2 Variants Including Multiple Mutants
Kwang-Eun Choi, Jeong-Min Kim, Jee Eun Rhee, Ae Kyung Park, Eun-Jin Kim, Cheon Kwon Yoo, Nam Sook Kang
International Journal of Molecular Sciences.2022; 23(9): 4956. CrossRef - SARS-CoV-2 shedding dynamics and transmission in immunosuppressed patients
Jee-Soo Lee, Ki Wook Yun, Hyeonju Jeong, Boram Kim, Man Jin Kim, Jae Hyeon Park, Ho Seob Shin, Hyeon Sae Oh, Hobin Sung, Myung Gi Song, Sung Im Cho, So Yeon Kim, Chang Kyung Kang, Pyoeng Gyun Choe, Wan Beom Park, Nam Joong Kim, Myoung-Don Oh, Eun Hwa Choi
Virulence.2022; 13(1): 1242. CrossRef - Immunological and Pathological Peculiarity of Severe Acute Respiratory Syndrome Coronavirus 2 Beta Variant
Sunhee Lee, Gun Young Yoon, Su Jin Lee, Young-Chan Kwon, Hyun Woo Moon, Yu-Jin Kim, Haesoo Kim, Wooseong Lee, Gi Uk Jeong, Chonsaeng Kim, Kyun-Do Kim, Seong-Jun Kim, Dae-Gyun Ahn, Miguel Angel Martinez
Microbiology Spectrum.2022;[Epub] CrossRef - Clinical scoring system to predict viable viral shedding in patients with COVID-19
Sung Woon Kang, Heedo Park, Ji Yeun Kim, Sunghee Park, So Yun Lim, Sohyun Lee, Joon-Yong Bae, Jeonghun Kim, Seongman Bae, Jiwon Jung, Min Jae Kim, Yong Pil Chong, Sang-Oh Lee, Sang-Ho Choi, Yang Soo Kim, Sung-Cheol Yun, Man-Seong Park, Sung-Han Kim
Journal of Clinical Virology.2022; 157: 105319. CrossRef - Model-informed COVID-19 exit strategy with projections of SARS-CoV-2 infections generated by variants in the Republic of Korea
Sung-mok Jung, Kyungmin Huh, Munkhzul Radnaabaatar, Jaehun Jung
BMC Public Health.2022;[Epub] CrossRef - Comparative analysis of mutational hotspots in the spike protein of SARS-CoV-2 isolates from different geographic origins
Sanghoo Lee, Mi-Kyeong Lee, Hyeongkyun Na, Jinwoo Ahn, Gayeon Hong, Youngkee Lee, Jimyeong Park, Yejin Kim, Yun-Tae Kim, Chang-Ki Kim, Hwan-Sub Lim, Kyoung-Ryul Lee
Gene Reports.2021; 23: 101100. CrossRef - Review of Current COVID-19 Diagnostics and Opportunities for Further Development
Yan Mardian, Herman Kosasih, Muhammad Karyana, Aaron Neal, Chuen-Yen Lau
Frontiers in Medicine.2021;[Epub] CrossRef - Locally harvested Covid-19 convalescent plasma could probably help combat the geographically determined SARS-CoV-2 viral variants
Manish Raturi, Anuradha Kusum, Mansi Kala, Garima Mittal, Anita Sharma, Naveen Bansal
Transfusion Clinique et Biologique.2021; 28(3): 300. CrossRef - Molecular Dynamics Studies on the Structural Characteristics for the Stability Prediction of SARS-CoV-2
Kwang-Eun Choi, Jeong-Min Kim, JeeEun Rhee, Ae Kyung Park, Eun-Jin Kim, Nam Sook Kang
International Journal of Molecular Sciences.2021; 22(16): 8714. CrossRef - Management following the first confirmed case of SARS-CoV-2 in a domestic cat associated with a massive outbreak in South Korea
Taewon Han, Boyeong Ryu, Suyeon Lee, Yugyeong Song, Yoongje Jeong, Ilhwan Kim, Jeongmin Kim, Eunjin Kim, Wonjun Lee, Hyunju Lee, Haekyoung Hwang
One Health.2021; 13: 100328. CrossRef - Genomic epidemiology reveals the reduction of the introduction and spread of SARS-CoV-2 after implementing control strategies in Republic of Korea, 2020
Jung-Hoon Kwon, Jeong-Min Kim, Dong-hun Lee, Ae Kyung Park, Il-Hwan Kim, Da-Won Kim, Ji-Yun Kim, Noori Lim, Kyeong-Yeon Cho, Heui Man Kim, Nam-Joo Lee, SangHee Woo, Chae Young Lee, Jin Sun No, Junyoung Kim, JeeEun Rhee, Myung-Guk Han, Gi-Eun Rhie, Cheon K
Virus Evolution.2021;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite