Articles
- Page Path
- HOME > Osong Public Health Res Perspect > Volume 13(1); 2022 > Article
-
Review Article
Points to consider for COVID-19 vaccine quality control and national lot release in Republic of Korea: focus on a viral vector platform -
Jung Hun Ju1
 , Naery Lee1, Sun-hee Kim1, Seokkee Chang1, Misook Yang1, Jihyun Shin1, Eunjo Lee1, Sunhwa Sung1, Jung-Hwan Kim1, Jin Tae Hong2, Ho Jung Oh1
, Naery Lee1, Sun-hee Kim1, Seokkee Chang1, Misook Yang1, Jihyun Shin1, Eunjo Lee1, Sunhwa Sung1, Jung-Hwan Kim1, Jin Tae Hong2, Ho Jung Oh1 -
Osong Public Health and Research Perspectives 2022;13(1):4-14.
DOI: https://doi.org/10.24171/j.phrp.2021.0311
Published online: February 8, 2022
1Emerging Infectious Disease Vaccines Division, National Institute of Food and Drug Safety Evaluation, Ministry of Food and Drug Safety, Cheongju, Korea
2College of Pharmacy and Medical Research Center, Chungbuk National University, Cheongju, Korea
- Corresponding author: Ho Jung Oh Emerging Infectious Disease Vaccines Division, National Institute of Food and Drug Safety Evaluation, Ministry of Food and Drug Safety, 187 Osongsaengmyeong 2-ro, Osong-eup, Heungdeok-gu, Cheongju 28159, Korea E-mail: ohojung@korea.kr
© 2022 Korea Disease Control and Prevention Agency.
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
- 5,733 Views
- 168 Download
Abstract
- Due to the global public health crisis caused by the coronavirus disease 2019 (COVID-19) pandemic, the importance of vaccine development has increased. In particular, a rapid supply of vaccines and prompt deployment of vaccination programs are essential to prevent and overcome the spread of COVID-19. As a part of the vaccine regulations, national lot release is regulated by the responsible authorities, and this process involves the assessment of the lot before a vaccine is marketed. A lot can be released for use when both summary protocol (SP) review and quality control testing are complete. Accelerated lot release is required to distribute COVID-19 vaccines in a timely manner. In order to expedite the process by simultaneously undertaking the verification of quality assessment and application for approval, it is necessary to prepare the test methods before marketing authorization. With the prolonged pandemic and controversies regarding the effectiveness of the COVID-19 vaccine against new variants, public interest for the development of a new vaccine are increasing. Domestic developers have raised the need to establish standard guidance on the requirements for developing COVID-19 vaccine. This paper presents considerations for quality control in the manufacturing process, test items, and SP content of viral vector vaccines.
- A common issue currently facing countries throughout the world is how to return to pre-coronavirus disease 2019 (COVID-19) daily life when the COVID-19 pandemic ends. COVID-19 is a respiratory syndrome caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which first emerged in Wuhan, Hubei Province, China in December 2019. In the absence of specific therapeutics or vaccines to prevent this new infectious disease, it has spread all over the world. Accordingly, the World Health Organization (WHO) declared the COVID-19 outbreak as a public health emergency of international concern on January 30, 2020. The WHO then made the assessment that COVID-19 could be characterized as a pandemic. At that time, about 120,000 people in 110 countries were infected with the virus [1,2].
- In the exceptional situation of a pandemic, the pharmaceutical industry, regulatory authorities, governments, and international organizations have coordinated collaborative strategies, and many national regulatory authorities have eased regulations to accelerate the development of COVID-19 vaccines. In order to facilitate access to COVID-19 vaccines, regulatory authorities have executed innovative and agile regulatory measures such as conditional marketing authorizations or emergency use authorizations. These measures are intended to ensure that patients could be supplied with COVID-19 therapeutics or vaccines as quickly as possible. These new regulatory applications involve expedited regulatory reviews for COVID-19 vaccines without compromising their safety, effectiveness, and quality [3–8]. As a result, the vaccine development process, which normally takes about 10 years, has been shortened to within about 1 year, from development to review and approval, and then to vaccination. Tables 1 and 2 show the authorization and approval status of COVID-19 vaccines by stringent regulatory authorities. In the Republic of Korea, the AstraZeneca vaccine, a viral vector platform vaccine was first authorized on February 10, 2021, followed by the Pfizer-BioNTech vaccine, Janssen vaccine, and Moderna vaccine, which have been approved and used [9].
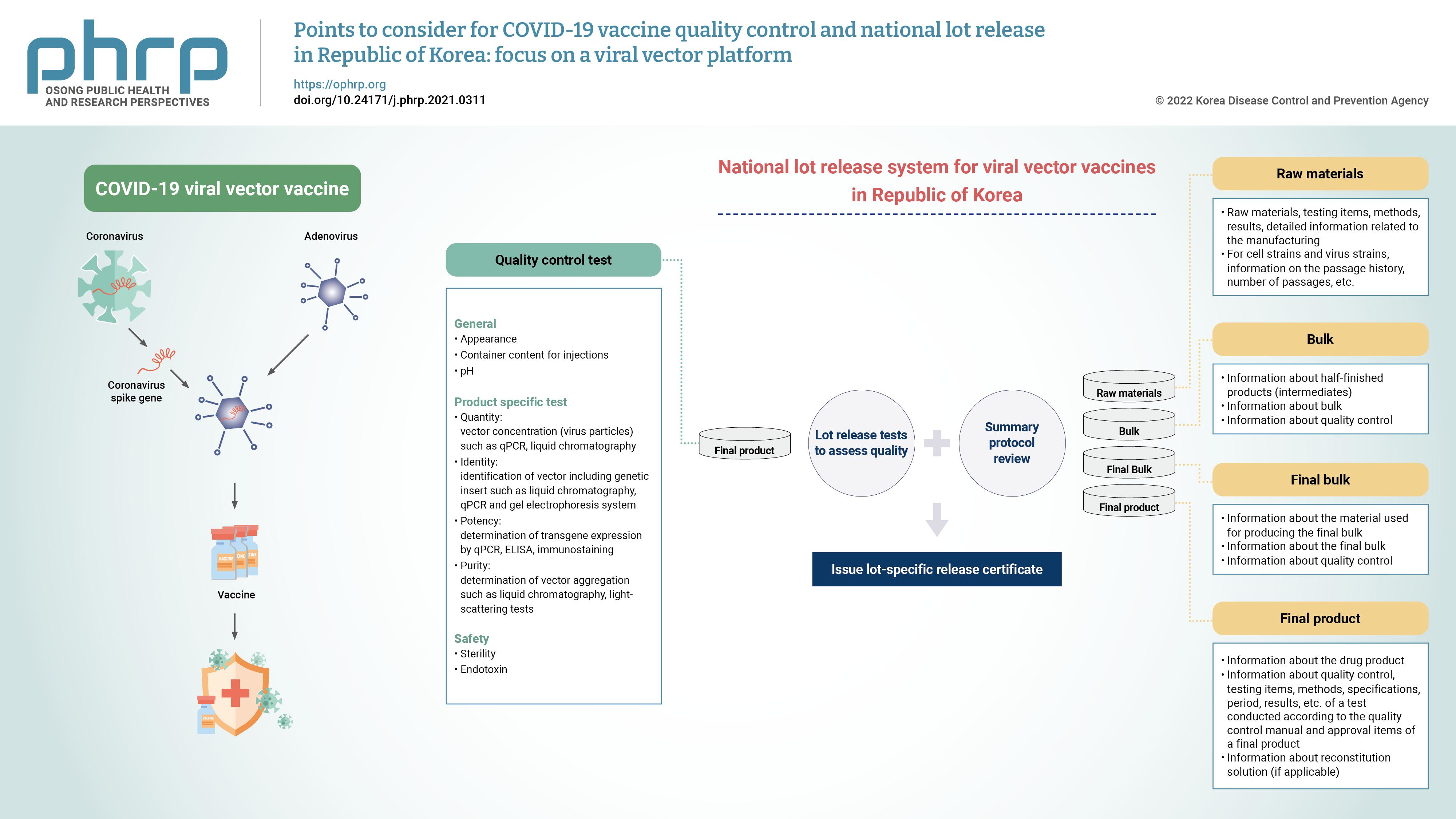
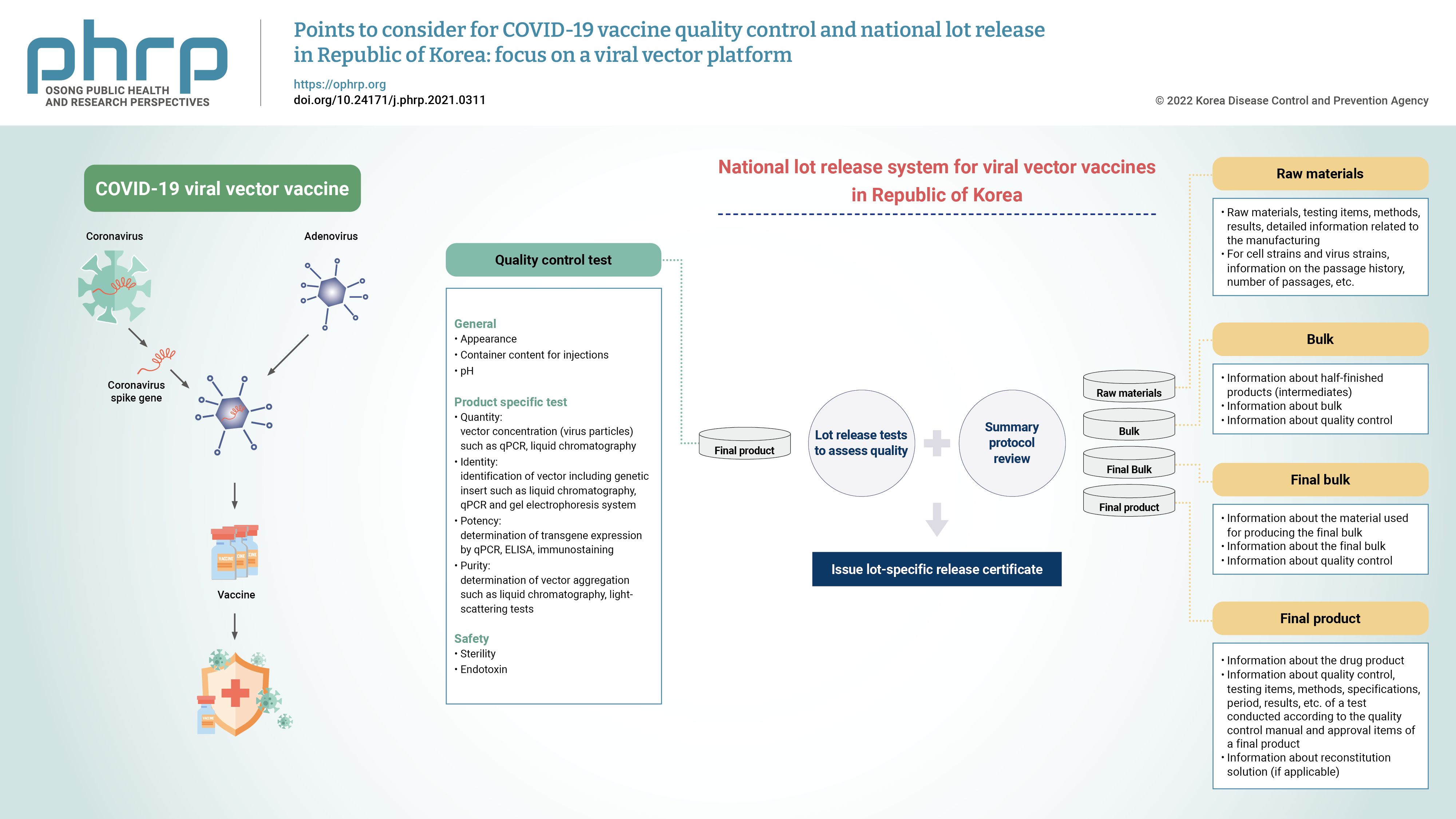
- In the Republic of Korea, it is stipulated that vaccines shall obtain lot release approval from the Ministry of Food and Drug Safety (MFDS) after undergoing the examination and verification of the data on manufacturing and quality management in accordance with the Pharmaceutical Affairs Act, Article 53. For biologics such as vaccines, reviews have been implemented to further confirm the quality of each lot of the product before its release onto the market. A national lot release system has been implemented to review the summary protocol (SP) for its production and quality control tests. The SP refers to a summarized document regarding the manufacturing process and test results of a product, ranging from raw materials to the final product (Figure 1). When the MFDS receives the application for a national lot release, the applicant is notified of the result after lot release testing and a review of the data concerning the product’s manufacturing and quality control. If the criteria are satisfied, a certificate of approval for the national lot release shall be issued [10]. Normally, the applicant is notified of the specifications and test methods of products for lot release, and then the subsequent lot release process is carried out after marketing authorization. However, as COVID-19 vaccines were designated as crisis-response medical products (specifically, as preventive vaccines for a pandemic infectious disease), they are subject to expedited national lot release. The MFDS has prioritized COVID-19 vaccines over other lot releases, and rapidly verified the quality of the vaccines [11]. The MFDS has committed to the improvement of the national quality control system for biologics and established a comprehensive lot release system. The National Institute of Food and Drug Safety Evaluation (NIFDS), an affiliated organization of the MFDS, is responsible for this task.
- To push ahead with the lot release further, simultaneous applications for marketing authorization and lot release have been conducted. The MFDS reviewed the safety and quality verification methods prior to the drug approval process of COVID-19 vaccines, and established the test methods before the application for lot release. Necessary equipment and instruments were also prepared. MFDS expedited the lot release process, which generally takes 2 or 3 months, and approved the lot release within 20 days. By the third quarter of 2021, the MFDS has approved the national lot release of 67.64 million doses (for domestic use) of COVID-19 vaccines. As of November 2021, 87.05 million doses—an amount with which more than 44 million people can be fully vaccinated—were approved for lot release. Thus, it became feasible to swiftly provide the necessary quantity of vaccines in a timely manner amid the spread of COVID-19. The status of domestic COVID-19 vaccine lot release and product information is available from the MFDS website (https://www.mfds.go.kr/vaccine_covid19.jsp) and the integrated drug information system (https://nedrug.mfds.go.kr), respectively.
- Vaccine manufacturing technology has been continuously developed as a solution to prevent infectious diseases. A vaccine platform refers to a technology for vaccine development by changing specific antigens or genetic information. A single vaccine platform commonly addresses a specific pathogen. However, the COVID-19 pandemic yielded a competitive situation, wherein various platforms were used to develop new vaccines with the goal of rapidly obtaining an effective vaccine. Currently, COVID-19 vaccine platform modalities include inactivated virus vaccines, attenuated virus vaccines, recombinant viral vector vaccines, recombinant subunit vaccines, DNA vaccines, and RNA vaccines [12,13]. As of December 3, 2021, there are 135 candidates in the clinical phase of development, of which vaccines using protein subunits accounted for 34.8% (n=47), RNA vaccines accounted for 15.6% (n=21), and viral vectors (non-replicating) accounted for 14.8% (n=20) (Table 3) [14].
- Since the manufacturing technology and quality control processes required for each platform are novel and unique, the establishment of test methods is time-consuming. Furthermore, each vaccine platform has advantages and drawbacks, including differences in production, efficacy, safety profile, and immune response. Therefore, it is essential to recognize the characteristics of every vaccine candidate and the specific quality considerations for each platform [12]. If new technology platform development guidelines are provided for use in quality control and testing, the vaccine development timeline could be significantly shortened. Accordingly, the NIFDS published 2 national lot release guidelines for new technology platforms for COVID-19 vaccines (viral vector vaccines and mRNA vaccines) by referring to the approval and review data of COVID-19 vaccines by the MFDS, WHO, and European Medicines Quality Committee (EDQM) guidelines [15,16].
- Viral vector vaccines are defined as live viruses that are genetically engineered to express 1 or more heterologous antigens [17]. Recombinant viral vector vaccines are generated by cloning the gene for an antigen from the pathogen into an avirulent host such as an adenovirus. Viral vector vaccines enter cells and produce the vaccine antigen, stimulating an immune response. Developing a recombinant viral vector vaccine that mimics a pathogen (e.g., SARS-CoV-2), which is the cause of the COVID-19 pandemic, but is not virulent, is known to be a safe option [12,13]. Several recombinant viral vectors derived from other viruses have emerged as gene delivery systems [18]. In particular, adenoviruses were initially characterized and have been used as gene delivery vectors since the early stages of gene therapy. Adenoviral vectors have been explored as vaccine agents for infectious diseases. Thus, as logical vaccine candidates, adenoviral vector vaccines are expected to be useful as a response to the COVID-19 pandemic as they are known to induce a potent and balanced immune response [19]. Two viral vector vaccines are currently commercialized in Republic of Korea. These vaccines contain adenovirus engineered to carry a SARS-CoV-2 gene, using a chimpanzee-derived adenovirus (AstraZeneca vaccine) or a human-derived adenovirus (Janssen vaccine) vector as a delivery system.
- In this paper, we describe considerations for the management of COVID-19 viral vector vaccines under the regulatory system in the Republic of Korea, with a particular focus on specific lot release tests and information to be included in the SP. This paper is intended to provide an internationally harmonized guideline by comparing and reviewing the EDQM document for recombinant viral vector vaccines [17], the WHO and Official Control Authority Batch Release (OCABR) guidelines [20–23], and the United States Pharmacopoeia (USP) COVID-19 vaccine quality assessment toolkits [24].
Introduction
- The quality control tests that can be performed at each stage of manufacturing a viral vector-based COVID-19 vaccine are as follows. This document is expected to help COVID-19 vaccine developers prepare an appropriate analytical strategy during vaccine development. Additional tests may be considered on a case-by-case basis, depending on the characteristics of the vector backbone and the specific manufacturing process established.
- Cell Substrates for Viral Vector Propagation
- The vector can be propagated in human diploid cell lines, continuous cell lines, chick-embryo cells, or in the amniotic membrane of chick embryos derived from specific pathogen-free chickens [17]. More specific test information on human diploid cells and immortal cell lines used as cell substrates for the propagation of recombinant viral vectors can be obtained from the MFDS guidelines [25,26] and European Pharmacopoeia Chapter 5.2.3 [27]. The cell substrates need to be used with a cell banking system.
- The most widely used method of viral vector development involves homologous recombination [28]. Certain replication-defective viral vectors may require testing for replication-competent viruses. Replication-competent viral vectors may incur significant issues with quality control when there is a wide homology region between the viral genome and the genome of the complementation cell. This expression could reduce the replication capacity by minimizing the homology between both genomes. It is recommended to use cells without homology between the vector and sequence for production.
- Viral Vector Seed Lot
- Recombinant viral vector production generally uses a seed lot system (master virus strain and working virus strain). The number of passages is controlled based on evidence regarding the maximum number of passages, and production cannot exceed the maximum passage level. Historical records, including information on the strain origin of the viral vector used for vaccine production, and in particular, subsequent manipulation of deleted or modified regions, should be confirmed. Genetic insert and flanking control regions should be described in detail, including the nucleotide sequences. The origin of genetic inserts into the vectors and the engineering methods should be documented. A relevant characterization could be carried out based on the nature of the viral vector and the results of pre-clinical studies.
- Propagation and Harvest
- It is preferable to produce vectors without the use of antibiotics. Penicillin or streptomycin should not be used at any stage of manufacture if insufficient data support doing so. It is necessary to provide information on control cell culture. A single harvest refers to the biological material prepared from a single production run.
- Bulk
- A bulk refers to the one containing active ingredient prior to formulation. The purification process could be applied to a pool of single harvests. The purification process should be validated for removal of impurities. The following test items may be considered for controlling the bulk: identification, vector concentration, infectious vector titer, ratio of vector particle concentration to infectious vector titer, ratio of infectious vector titer to total protein concentration, transgene expression, host-cell protein, host-cell DNA, vector aggregation, residual reagents, residual antibiotics, the absence of replication-competent viral vectors, microbial control (microbial limit or sterility), and endotoxin.
- Final Bulk
- The final bulk may consist of 1 or more bulks. In preparing the final bulk, any excipients such as stabilizers are added to the product. The following tests could be considered for control of the final bulk: microbial control (microbial limit or sterility) and preservative.
- Final Product
- Control tests for the final product are shown in Table 4. If a test for bovine serum albumin or ovalbumin (if applicable) has been carried out on the bulk or the final bulk, they may be omitted on the final product.
Considerations for Quality Control in Each Stage of the Production of Viral Vector Vaccines
- Test Items and Methods
- Regulatory authorities in the countries such as the United States, Europe, and Japan are supporting the development of COVID-19 vaccines by providing guidance, quality standards, and training materials to vaccine developers and manufacturers [20–24]. In the context of the COVID-19 pandemic, the EDQM published the first analytical strategy options on the quality control of COVID-19 recombinant viral vector vaccines in November 2020. In this document, the mandatory tests at each stage of viral vector vaccine manufacturing (from raw materials to the final product) are presented [20]. The USP provided a toolkit for the quality evaluation items and test methods for each COVID-19 vaccine platform (mRNA vaccine, inactivated vaccine, and viral vector vaccine) in July 2021 [24]. Japan registered a monograph of test items and standards for domestically authorized COVID-19 vaccines (Pfizer-BioNTech, Moderna, and AstraZeneca) in the Minimum Requirements for Biological Products (MRBP) [22]. The NIFDS published a Guideline on National Lot Release for COVID-19 Viral Vector Vaccines [15], and SARS-CoV-2 viral vector vaccines fee for national lot release was added to the Regulation on Fees for Pharmaceutical Approval, etc. on October 21, 2021 [29]. In addition, in order to ensure the transparency and clarify of the lot release system, revisions of the Regulation for Designation, Approval Process, and Method of Pharmaceuticals for National Lot Release, which describes the amount of samples and test items for newly authorized COVID-19 vaccines, are in progress, and the proposed amendment will be announced within this year.
- The test items, objectives, and possible methods in Table 4 are necessary information to be considered in the quality control of the final product, but they are not set criteria for lot release. Generally, test specifications and methods subject to lot release should be in compliance with the MRBP (NFDS Notification). However, the Minister of the MFDS may adjust them partially considering the characteristics of the product according to the approved Specification and Analytical Procedures.
- National Lot Release Tests
- Specifications and test methods include product-specific tests such as identification, purity, potency, and tests for preservatives and stabilizers used in manufacturing. Further, sterility and endotoxin tests are conducted to confirm product safety. In addition, in accordance with the MRBP (MFDS Notification), the details of the necessary criteria such as the nature, condition, and quality of vaccines could be determined. Table 5 demonstrates the recommended test items and methods for viral vector vaccines. The test items performed for the national lot release of viral vector vaccines (AstraZeneca and Janssen vaccines) authorized in Korea are as follows. The lot release test items of AstraZeneca vaccine include appearance, pH, sterility, endotoxin, identity, potency (infectivity), virus particle concentration, purity (ratio of DNA to protein, ratio of virus particle to infectious virus), and container content for injections. The test items for Janssen vaccine lot release include appearance, pH, sterility, endotoxin, identity (virus identity, virus protein identity), potency (transgene expression, infectious titer, and ratio of virus particle to infectious virus), concentration, purity, and container content for injections. The test items for both vaccines are included in the revised content of the Regulation for Designation, Approval Process, and Method of Pharmaceuticals for National Lot Release.
- Table 6 shows the test items for COVID-19 viral vector vaccines tested by the National Regulatory Authority and National Control Laboratory. The test items performed by the European Network of Official Medicines Control Laboratories are specified in the OCABR guideline [21], and the Australian government provides guidance on the assessment items for COVID-19 vaccine batch release through the Therapeutic Goods Administration website [23].
- SP Review
- The manufacturer’s SP is the summarized information taken from the manufacturing and testing results according to Good Manufacturing Practice requirements to ensure that the lot meets the specifications of the marketing authorization. The SP contains data on all appropriate production steps and controls, and it is certified and signed by the person in charge of the manufacturing company [30].
- As shown in Table 7, information on the production and storage at each stage of the manufacturing process, including information on the passage history of cell strains/virus strains used as raw materials, as well as the test methods, specifications, and results should be provided in the SP. For reference, an SP template is included with the Guideline on National Lot Release for COVID-19 Viral Vector Vaccines [15]. The manufacturer or importer will be notified of the SP form. However, it is thus possible that SP for a specific product may differ in detail from the provided model.
Guidance on Quality Control for Viral Vector Vaccines
- The still ongoing COVID-19 pandemic demands a global response due to the occurrence and spread of variants with novel mutations. Around the world, people are looking for explicit scientific evidence and useful guidance to deal with this unprecedented pandemic. The currently predominating Delta variant lowers the effectiveness of vaccination, leading to the 4th wave of the epidemic [31]. The Delta variant is eightfold less sensitive to ChAdOx1 vaccine-elicited antibodies than the wild-type Wuhan-1. It was also confirmed that the neutralizing antibodies from those who had recovered or been vaccinated showed decreased effectiveness against the Delta variant [32]. Although the current vaccines could reduce the hospitalization and mortality rate, it is also necessary to develop a next-generation vaccine to cope with breakthrough infections. Vaccination is recognized as the best game-changer that mitigates the current pandemic [33], considering its superior effectiveness and capability as a safe public health intervention to prevent infections. Therefore, the rapid production and supply of safe and effective vaccines to protect people and mitigate the economic and social impacts of infectious diseases should be prioritized [34,35].
- In the middle of the 20th century, vaccine production technology made substantial advances as vaccines were produced by virus propagation in cell culture. Through these technological innovations, vaccine development platforms have been diversified [12]. Historically, 4 classic platforms (inactivated virus, live-attenuated virus, protein subunit, and virus-like particle) constituted most vaccine products [13]. However, in response to COVID-19, developers are looking for more up-to-date technologies in order to avoid the safety and efficacy concerns associated with traditional platforms targeting SARS-CoV-2 [12]. The currently commercialized COVID-19 vaccines were developed quickly, even though they were based on recent technologies. The reasons for the success of vaccine development with a rapid timeline are as follows: SARS-CoV-2 is closely related to SARS-CoV, and through the accumulated data on SARS-CoV and Middle East respiratory syndrome coronavirus, a spike protein has been identified as the antigenic target for coronavirus vaccines [36,37]. This enabled the more rapid design and development of SARS-CoV-2 vaccine candidates after the emergence of this new virus, as well as sharing the SARS-CoV-2 gene sequence, diversifying vaccine development strategies, supporting development costs, and shortening the period from vaccine approval to distribution by expediting the regulatory response system to COVID-19. In other words, the rapid development and deployment of COVID-19 vaccines resulted from organically harmonized efforts and collaboration to manage this public health crisis.
- One of the hurdles regarding COVID-19 vaccine development is how to decide upon the most appropriate platform to target SARS-CoV-2. It has not been determined which kind of platform would be the most effective against the novel coronavirus. Therefore, all vaccine platforms are being explored as COVID-19 vaccine candidates. Inactivated, attenuated, and recombinant vaccines using pathogens or antigenic proteins have been successful for targeting other infectious diseases. Novel platforms such as viral vector vaccines or nucleic acid vaccines have not been sufficiently used to fully establish their safety and efficacy in humans, but these platforms have been used worldwide as major vaccines for COVID-19. There are 135 candidates in the clinical development stage, of which viral vector vaccines and nucleic acid vaccines account for about 43.0% (36 nucleic acid platforms and 22 viral vector platforms) as of December 3, 2021 (Table 3).
- Unlike chemically synthesized pharmaceuticals, vaccines are manufactured from biological sources. They should be produced consistently and show comparability from lot to lot, demonstrating clinical efficacy, immunogenicity, and safety. Furthermore, since vaccines are biological products with a complex nature and inherent potential for variability, it is difficult to produce vaccines with consistency, safety, and effectiveness even under similar manufacturing conditions. Therefore, an independent review of manufacturing and quality control data from each vaccine lot is essential to assure the consistent quality of manufactured lots [30]. Reliable and authorized guidance needs to provide support through regulatory requirements for quality management. Since the outbreak of the COVID-19 pandemic, regulatory agencies such as the WHO, Europe, United States, and Japan have provided high-quality documents in order to support COVID-19 vaccine developers currently working on candidate vaccines based on recent technologies, such as nucleic acid vaccines and recombinant viral vector vaccines. The Republic of Korea has published lot release guidelines on COVID-19 vaccine platforms (viral vector vaccines, mRNA vaccines) to harmonize the global regulatory requirements for lot release and to accelerate vaccine development [15,16].
- This paper is intended to serve as guidance for regulatory systems dealing with the lot release. Furthermore, this paper also provides general considerations for quality control and tests applicable to vaccine development and quality evaluation using the viral vector platform. Lot release test items and methods provided are prepared based on drug approval and review cases, and national lot release cases for domestically approved replication-defective adenoviral vector vaccines. This information will help vaccine developers to minimize industry trials and expedite commercialization by providing predictable regulatory requirements for the same platform vaccine. However, it should be noted that quality control items and considerations may be different depending on the replication-competent viral vector and/or the nature of the vector backbone as the vaccine delivery system. The quality tests provided herein have been drawn up using available knowledge to date, and the content may be updated as needed to change the regulatory system and/or to reflect further experience gained with new products. Emerging infectious disease disasters such as the COVID-19 pandemic are expected to occur periodically. In order to accelerate vaccine development for novel infectious diseases such as COVID-19, technical support for globally harmonized quality requirements is urgently needed.
Conclusion
-
Ethics Approval
Not applicable.
-
Conflicts of Interest
The authors have no conflicts of interest to declare.
-
Funding
None.
-
Availability of Data
All data generated or analyzed during this study are included in this published article. Other data may be requested from the corresponding author.
-
Authors’ Contributions
Conceptualization: JHJ, JTH; Data curation: NL, SHK, SC, MY, JS, EL, SS, JHK; Writing-original draft: JHJ, NL, HJO; Writing-review & editing: JTH, HJO.
Article information

Available from regulatory authority websites as of October 2021 (sources: FDA, https://www.fda.gov/emergency-preparedness-and-response/counterterrorism-and-emerging-threats/coronavirus-disease-2019-covid-19; EMA, https://www.ema.europa.eu/en; PMDA, https://www.pmda.go.jp/english/about-pmda/0002.html; Health Canada, https://www.canada.ca/en/health-canada/services/drugs-health-products/covid19-industry/drugs-vaccines-treatments/vaccines.html; Swissmedic, https://www.swissmedic.ch/swissmedic/en/home.html).
COVID-19, coronavirus disease 2019; ICH, International Council for Harmonization; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; FDA, Food and Drug Administration; MA, marketing authorization; EUA, emergency use authorization; EC, European Commission; CMA, conditional marketing authorization; MHLW, Ministry of Health, Labour and Welfare; PMDA, Pharmaceuticals and Medical Devices Agency; SAFE, special approval for emergency; HC, Health Canada; ISAD IO, interim order pathway for COVID-19-related drugs and vaccines; TPA, temporary authorization; -, unauthorized or unapproved.
COVID-19, coronavirus disease 2019; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Available from Ministry of Food and Drug Safety website (source: https://nedrug.mfds.go.kr).
COVID-19, coronavirus disease 2019.
Data as of December 3, 2021 (source: https://www.who.int).
|
Category |
United States (USP) |
Europe (EDQM) |
Korea (MFDS) |
Japan (NIID)a) |
|
|---|---|---|---|---|---|
| Item | Attribute | Possible test methods | |||
| Identity | Sequence confirmation | DNA extraction & sequencing (USP 1125, 1126) restriction analysis (USP 1129, 1126) | Immunochemical methods (EP 2.7.1), NAT (EP 2.6.21), LC (EP 2.2.29) | Immunochemical methods. NAT, LC | Suitable methods |
| Vector detection | qPCR, ddPCR, RT-PCR (USP 1125, 1126, 1127), ELISA (USP 1103) | ||||
| Purity | Vector aggregates | Light-scattering (USP 1430.2, 1430.3, 1430.5, 1430.6, 1430.7), SEC-MALS (USP 621, 1430.1) | Light-scattering | Light-scattering | - |
| Potency | Infectious vector titer | Plaque assays (USP 111, 1235, 1237), CCID50, cell-based qPCR (USP 1032, 1033, 1034) | Plaque assays or CCID50 assay by immunostaining, qPCR, flowcytometry/FACS, fluorescent focus assay | Plaque assays or CCID50 assay by immunostaining, qPCR, flowcytometry, fluorescent focus assay | Immunostaining |
| Transgene expression | Western blot (USP 1104), ELISA (USP 1103), LC-MS (USP 621, 736, 1736), RP-HPLC (USP 621) | Immunochemical assay (EP 2.7.1), biochemical assay, flow cytometry (EP 2.7.24) | Immunochemical assay, biochemical assay, flow cytometry | - | |
| Quantity | Virus particle (vector concentration) | Light-scattering & DLS (USP 1430.2, 1430.6), CZE (USP 1053), qPCR (USP 1125, 1126, 1127) | qPCR (EP 2.6.21) | qPCR | LC |
| Appearance | Compendial test | USP 1, 790 | EP 2.7.1, 2.6.21, 2.2.29 | Visual observation | Criteria specified |
| pH | Compendial test | USP 791 | EP 2.2.3 | KP general test | JP general test |
| Container content for injections (includes extractable volume) | Compendial test | USP 697 | EP 2.9.17 | KP general test | - |
| Sterility | Compendial test | USP 71 | EP 2.6.1 | KP general test | JP general test |
| Endotoxin | Compendial test | USP 85 | EP 2.6.14 | KP general test | JP general test |
| Osmolality | Compendial test | USP 785 | EP 2.2.35 | KP general test | - |
USP, United States Pharmacopoeia; EDQM, European Medicines Quality Committee; MFDS, Ministry of Food and Drug Safety; NIID, National Institute of Infectious Diseases; EP, European Pharmacopoeia; NAT, nucleic acid test; LC, liquid chromatography; qPCR, quantitative polymerase chain reaction; ddPCR, droplet digital PCR; RT-PCR, reverse transcription PCR; ELISA, enzyme-linked immunosorbent assay; SEC, size exclusion chromatography; MALS, multi-angle light-scattering; CCID50, cell culture infectious dose 50%; FACS, fluorescence-activated cell sorting; MS, mass spectrometry; DLS, dynamic light-scattering; CZE, capillary zone electrophoresis; RP, reverse phase; HPLC, high-performance liquid chromatography; KP, The Korean Pharmacopoeia; JP, Japanese Pharmacopoeia; -, not applicable.
a) Chimpanzee adenoviral vector vaccine only.
- 1. World Health Organization (WHO). Coronavirus disease 2019 (COVID-19) Situation report - 51 [Internet]. Geneva: WHO; 2020 [cited 2022 Jan 8]. Available from: https://www.who.int/publications/m/item/situation-report---51.
- 2. World Health Organization (WHO). A year without precedent: WHO’S COVID-19 response [Internet]. Geneva: WHO; 2020 [cited 2022 Jan 8]. Available from: https://www.who.int/news-room/spotlight/a-year-without-precedent-who-s-covid-19-response/.
- 3. World Health Organization (WHO). Considerations for evaluation of COVID19 vaccine, ver. 25 [Internet]. Geneva: WHO; 2020 [cited 2022 Jan 8]. Available from: https://www.who.int/publications/m/item/considerations-for-the-assessment-of-covid-19-vaccines-for-listing-by-who.
- 4. U.S. Food and Drug Administration (FDA). Emergency use authorization for vaccines to prevent COVID-19 guidance for industry: guidance for industry [Internet]. Silver Spring, MD: FDA; 2021 [cited 2022 Jan 8]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/emergency-use-authorization-vaccines-prevent-covid-19.
- 5. Cavaleri M, Enzmann H, Straus S, et al. The European Medicines Agency's EU conditional marketing authorisations for COVID-19 vaccines. Lancet 2021;397:355−7.ArticlePubMedPMC
- 6. Maeda H. Japan's Special Approval for Emergency system during the COVID-19 pandemic. Clin Pharmacol Ther 2021 May 25 [Epub];https://doi.org/10.1002/cpt.2310.Article
- 7. Health Canada. Guidance for market authorization requirements for COVID-19 vaccines: overview [Internet]. Ottawa: Health Canada; 2021 [cited 2022 Jan 8]. Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/covid19-industry/drugs-vaccines-treatments/guidance-market-authorization-vaccines.html.
- 8. Swiss Agency for Therapeutic Products (Swissmedic). Guidance document authorisation procedures for COVID-19 medicinal products during a pandemic HMV4 [Internet]. [cited 2021 May 15]. Available from: https://www.swissmedic.ch›zulassung›zl_hmv_iv.
- 9. Ministry of Food and Drug Safety (MFDS). Latest COVID-19 News [Internet]. Cheongju: MFDS; 2021 [cited 2022 Jan 8]. Available from: https://www.mfds.go.kr/eng/brd/m_64/list.do.
- 10. Ministry of Food and Drug Safety (MFDS). Regulation on safety of pharmaceuticals, etc. [Internet]. Cheongju: MFDS; 2021 [cited 2022 Jan 8]. Available from: https://www.mfds.go.kr/eng/brd/m_18/view.do?seq=71487.
- 11. Ministry of Food and Drug Safety (MFDS). Regulation for designation, approval process, and Method of Pharmaceuticals for National Lot Release [Internet]. Cheongju: MFDS; 2021 [cited 2022 Jan 8]. Available from: https://www.mfds.go.kr/brd/m_211/view.do?seq=14580. Korean.
- 12. Verdecia M, Kokai-Kun JF, Kibbey M, et al. COVID-19 vaccine platforms: delivering on a promise? Hum Vaccin Immunother 2021;17:2873−93.ArticlePubMedPMC
- 13. van Riel D, de Wit E. Next-generation vaccine platforms for COVID-19. Nat Mater 2020;19:810−2.ArticlePubMed
- 14. World Health Organization (WHO). COVID-19 vaccine tracker and landscape [Internet]. Geneva: WHO; 2022 [cited 2022 Jan 8]. Available from: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines.
- 15. Ministry of Food and Drug Safety (MFDS), National Institute of Food and Drug Safety Evaluation (NIFDS). Guideline on national lot release for COVID-19 viral vector vaccine [Internet]. Cheongju: MFDS; 2021 [cited 2022 Jan 8]. Available from: https://www.mfds.go.kr/brd/m_1060/view.do?seq=14863&srchFr=&srchTo=&srchWord=%EC%B6%9C%ED%95%98&srchTp=0&itm_seq_1=0&itm_seq_2=0&multi_itm_seq=0&company_cd=&comcomp_nm=&Data_stts_gubun=C9999&page=1. Korean.
- 16. Ministry of Food and Drug Safety (MFDS), National Institute of Food and Drug Safety Evaluation (NIFDS). Guideline on national lot release for COVID-19 mRNA vaccine [Internet]. Cheongju: MFDS; 2021 [cited 2022 Jan 8]. Available from: https://www.mfds.go.kr/brd/m_1060/view.do?seq=14864&srchFr=&srchTo=&srchWord=%EC%B6%9C%ED%95%98&srchTp=0&itm_seq_1=0&itm_seq_2=0&multi_itm_seq=0&company_cd=&comcomp_nm=&Data_stts_gubun=C9999&page=1. Korean.
- 17. European Directorate for the Quality of Medicines and HealthCare (EDQM). Recombinant viral vectored vaccines for human use [Internet]. Strasbourg: EDQM; 2020 [cited 2022 Jan 8]. Available from: https://www.edqm.eu/sites/default/files/medias/fichiers/COVID-19/recombinant_viral_vectored_vaccines.pdf.
- 18. Condit RC, Williamson AL, Sheets R, et al. Unique safety issues associated with virus-vectored vaccines: Potential for and theoretical consequences of recombination with wild type virus strains. Vaccine 2016;34:6610−6.ArticlePubMedPMC
- 19. Mendonca SA, Lorincz R, Boucher P, et al. Adenoviral vector vaccine platforms in the SARS-CoV-2 pandemic. NPJ Vaccines 2021;6:97. ArticlePubMedPMC
- 20. World Health Organization (WHO). WHO operational tool for efficient and effective lot release of SARS-CoV-2 (Covid-19) vaccine version 1 [Internet]. Geneva: WHO; 2021 [cited 2022 Jan 8]. Available from: https://extranet.who.int/pqweb/sites/default/files/documents/WHO_OperationalTool_EfficientLotRelease_v20Jan2021.pdf.
- 21. European Directorate for the Quality of Medicines and HealthCare (EDQM). Guideline for pandemic COVID-19 vaccine (Non-replicating adenovirus- vectored vaccine) [Internet]. Strasbourg: EDQM; 2021 [cited 2022 Jan 8]. Available from: https://www.edqm.eu/en/ocabr-activities-related-covid-19-vaccines.
- 22. National Institute of Infectious Diseases, Japan (NIID). Minimum requirements for biological product: COVID-19 (SARS-CoV-2) vaccine (recombinant chimpanzee adenovirus vector) [Internet]. Tokyo: NIID; 2021 [cited 2021 Jul 30]. Available from: https://www.niid.go.jp/niid/ja/mrbp.html.
- 23. Therapeutic Goods Administration (TGA). Batch release assessment of COVID-19 vaccines [Internet]. Woden: Department of Health Therapeutic Goods Administration, Australia; 2022 [cited 2022 Jan 8]. Available from: https://www.tga.gov.au/batch-release-assessment-covid-19-vaccines.
- 24. United States Pharmacopeia (USP). USP COVID-19 vaccine quality assessment toolkits [Internet]. Rockville, MD: USP; 2021 [cited 2022 Jan 8]. Available from: https://www.usp.org/covid-19/quality-attributes-toolkits.
- 25. Ministry of Food and Drug Safety (MFDS), National Institute of Food and Drug Safety Evaluation (NIFDS). Guidance on the characterization of cell substrates used to produce biologicals [Internet]. Cheongju: MFDS; 2010 [cited 2022 Jan 8]. Available from: https://www.mfds.go.kr/brd/m_1060/view.do?seq=12951&srchFr=&srchTo=&srchWord=%ED%99%94%EC%9E%A5%ED%92%88&srchTp=0&itm_seq_1=0&itm_seq_2=0&multi_itm_seq=0&companc_cd=&company_nm=&Data_stts_gubun=C9999&page=3. Korean.
- 26. Ministry of Food and Drug Safety (MFDS), National Institute of Food and Drug Safety Evaluation (NIFDS). Guidance on the testing for adventitious agents of biological products for human use [Internet]. Cheongju: MFDS; 2010 [cited 2022 Jan 8]. Available from: https://www.mfds.go.kr/brd/m_1060/view.do?seq=12952&srchFr=&srchTo=&srchWord=&srchTp=&itm_seq_1=0&itm_seq_2=0&multi_itm_seq=0&company_cd=&company_nm=&page=83. Korean.
- 27. European Pharmacopoeia Online 10.0. 5.2.3 Cell substrate for the production of vaccine for human use [Internet]. Strasbourg: European Directorate for the Quality of Medicines and HealthCare; 2021 [cited 2022 Jan 8]. Available from: https://pheur.edqm.eu/internal/fe4bccca68c1481682fc276306dba666/10-5/10-5/page/50203E.pdf.
- 28. Zhang W, Fu J, Ehrhardt A. Novel vector construction based on alternative adenovirus types via homologous recombination. Hum Gene Ther Methods 2018;29:124−34.ArticlePubMed
- 29. Ministry of Food and Drug Safety (MFDS). Regulation on fees for pharmaceutical approval, etc. [Internet]. Cheongju: MFDS; 2020 [cited 2022 Jan 8]. Available from: https://www.mfds.go.kr/brd/m_211/view.do?seq=14537. Korean.
- 30. Ministry of Food and Drug Safety (MFDS), National Institute of Food and Drug Safety Evaluation (NIFDS). Guideline on national lot release [Internet]. Cheongju: MFDS; 2016 [cited 2022 Jan 8]. Available from: https://www.mfds.go.kr/eng/brd/m_27/view.do?seq=70903.
- 31. Lee JK. How to deal with the Delta variant this fall. Osong Public Health Res Perspect 2021;12:201−2.ArticlePubMedPMC
- 32. Mlcochova P, Kemp SA, Dhar MS, et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 2021;599:114−9.PubMedPMC
- 33. Yoo JH. What we do know and do not yet know about COVID-19 vaccines as of the beginning of the year 2021. J Korean Med Sci 2021;36:e54.ArticlePubMedPMC
- 34. Andre FE, Booy R, Bock HL, et al. Vaccination greatly reduces disease, disability, death and inequity worldwide. Bull World Health Organ 2008;86:140−6.ArticlePubMed
- 35. Nagy A, Alhatlani B. An overview of current COVID-19 vaccine platforms. Comput Struct Biotechnol J 2021;19:2508−17.ArticlePubMedPMC
- 36. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270−3.PubMedPMC
- 37. Yong CY, Ong HK, Yeap SK, et al. Recent advances in the vaccine development against Middle East respiratory syndrome-coronavirus. Front Microbiol 2019;10:1781. ArticlePubMedPMC
 PubReader
PubReader ePub Link
ePub Link Cite
Cite